This blog was written by guest author Michael Curtin, Senior Product Manager, Small Molecule Drug Discovery.
ATP is the universal energy currency of cells, and thousands of proteins outside the kinase family “spend” it to move cargo, remodel nucleic acids, pump ions, or fold proteins. These ATP-hydrolyzing enzymes—collectively known as ATPases—span functional classes including motor proteins, transporters, chaperones, chromatin remodelers, ligases, and, crucially for genome stability, helicases.
From DNA replication to RNA processing, helicases are essential players. DNA/RNA helicases such as MCM, XPB/XPD, WRN, and members of the DDX family sit alongside AAA+ unfoldases, ABC transporters, and V-ATPases—all drawing on ATP to power their molecular work.
In April 2024, Promega hosted the “Target Engagement in Chemical Biology Symposium” at the Kornberg Center, a research and development hub on Promega’s campus in Madison, Wisconsin. The goal of the symposium was to gather interdisciplinary researchers interested in the field of small molecule target engagement to foster collaboration through knowledge sharing and innovation. The symposium featured a 1.5-day agenda packed with 23 speakers, 4 workshops, poster sessions and social events. Over 130 attendees gathered to participate in the multifaceted event, with participants from different geographic regions and in different research sectors from academia to government to industry.
Attendees gather for the poster session in Kornberg Atrium. Photo by Anna Bennett (Promega Corporation)
The symposium highlighted the collective commitment to overcoming the challenges in drug discovery by developing more targeted and efficacious treatments, driven by a shared determination to create innovative solutions that address unmet medical needs. While we cannot share all the exciting research presented at the symposium, we are thrilled to highlight a few talks that exemplify the novel work and collaborative spirit of this research community.
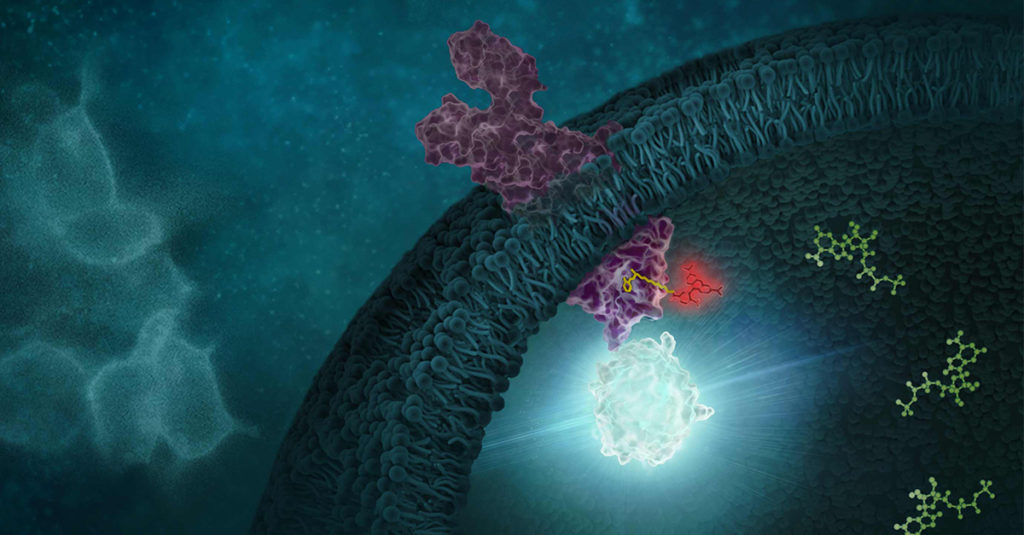
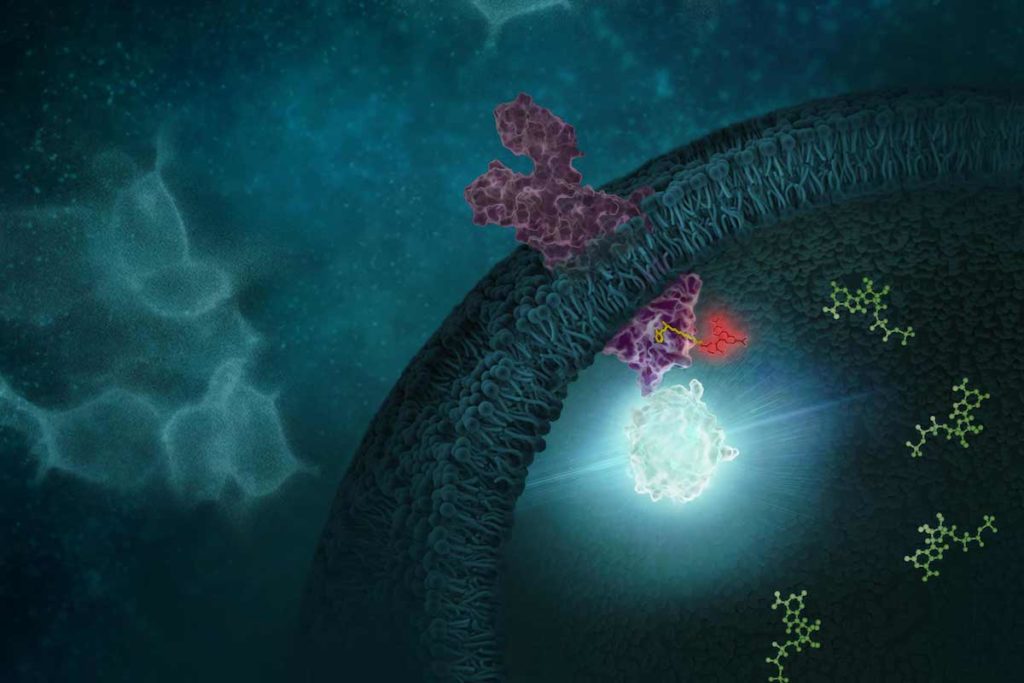
Identifying Inflammasome Inhibitors: What’s Missing The NLRP3 inflammasome is implicated in a wide range of diseases. The ability to inhibit this protein complex could provide more precise, targeted relief to inflammatory disease sufferers than current broad-spectrum anti-inflammatory compounds, potentially without side effects.
Studies of NLRP3 inflammasome inhibitors have relied on cell-free assays using purified NLRP3. But cell-free assays cannot assess physical engagement of the inhibitor and target in the cellular micro-environment. Cell-free assays cannot show if an NLRP3 inhibitor enters the cell, binds the target and how long the inhibitor binding lasts.
Cell-based assays that interrogate the physical interaction of the NLRP3 target and inhibitor inside cells are needed.
Image adapted from original artwork by iSO-FORM LLC.
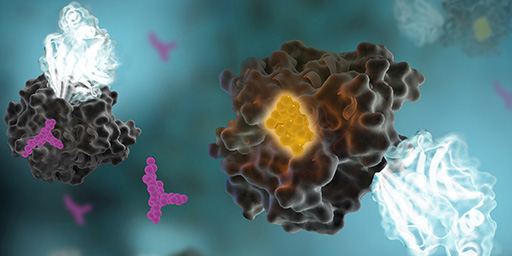
We made the cover! Of Cell Chemical Biology, that is.
This July, Cell Chemical Biology editors accepted a study from Promega scientists and invited the research team to submit cover art for the issue. The study in question details a BRET-based method to quantify drug-target occupancy within RAF-KRAS complexes in live cells. Promega scientists Matt Robers and Jim Vasta collaborated with one of our talented designers, Michael Stormberg, to craft an image that accurately represents the science in a dynamic and engaging way.
I spoke with Michael Stormberg to learn more about the creative process that went into creating this cover art and how he worked with the research team and other collaborators.
Mitogen-activated protein kinases (MAPKs) are a large family of proteins that regulate diverse cellular functions in eukaryotes, including gene expression, proliferation, differentiation and apoptosis (1). MAPK signaling pathways typically include three sequentially activated kinases, and these pathways are triggered in response to extracellular stimuli, such as cytokines, mitogens, growth factors and oxidative stress (1). Ultimately, the signal is transmitted to the nucleus, with the activation of a specific transcription factor that modulates the expression of one or more genes.
Among MAPK pathways, the RAS-RAF-MEK-ERK signaling pathway has been studied extensively. Mutations in RAS family proteins and resultant dysregulation of the signaling pathway are implicated in a variety of cancers. Therefore, this pathway is a popular target for anticancer drug development.
Monitoring and quantifying drug-target binding in a live-cell setting is important to bridging the gap between in vitro assay results and the phenotypic outcome, and therefore represents a crucial step in target validation and drug development (1). The NanoBRET™ Target Engagement (TE) assay is a biophysical technique that enables quantitative assessment of small molecule-target protein binding in live cells. This live-cell target engagement assay uses the bioluminescence resonance energy transfer (BRET) from a NanoLuc® luciferase-tagged target protein and a cell-permeable fluorescent tracer that reversibly binds the target protein of interest. In the presence of unlabeled test compound that engages the target protein, the tracer is displaced, and a loss of BRET signal is observed. Due to the tight distance constraints for BRET, the signal measured is specific to the target fused to NanoLuc® luciferase.
Promega offers over 400 ready-to-use assays for multiple target classes, including kinases, E3 ligases, RAS, and many others. For targets that do not have an existing NanoBRET™ TE assay, Promega offers NanoBRET™ dyes, NanoLuc® cloning vectors, and NanoBRET™ detection reagents to develop novel NanoBRET™ TE assays.
One critical component in the development of novel NanoBRET™ TE assay is the creation of the cell-permeable fluorescent tracers (NanoBRET™ tracers) against the target protein of interest. The tracers are bifunctional, consisting of a NanoBRET™-compatible fluorophore and a target-binding moiety connected by a linker. While the NanoBRET™ 590 dyes have demonstrated consistently robust cell permeability and optimal spectral overlap with NanoLuc® for BRET, a ligand capable of binding to the target protein of interest needs to be identified to generate a NanoBRET™ tracer.
What Are DNA-Encoded Libraries?
DNA-Encoded Libraries, (DELs), have emerged as powerful tools for discovering small molecule ligands to target proteins of interest at an unprecedented scale. . owing to the ability of a DEL to enable the synthesis of larger libraries of compounds and to target proteins without any prior structural knowledge of the proteins or their ligands (2). Because each member of a DEL contains a DNA barcode and a small molecule separated by a linker, DEL is primed for discovering leads within therapeutic modalities that rely on bifunctional chemistry, such as proteolysis targeting chimeras (PROTACs). Since NanoBRET™ tracers are also bifunctional, ligands identified from DEL selections could serve as ideal candidates for developing novel NanoBRET™ tracers that can enable NanoBRET™ TE assays for new targets.
G protein-coupled receptors (GPCRs) comprise a large group of cell surface receptors, characterized by the unique structural property of crossing the cell membrane seven times. They respond to a diverse group of signaling molecules, such as peptides, neurotransmitters, cytokines, hormones and other small molecules (1). Upon activation, GPCRs interact with GTP-binding (G) proteins and arrestins to regulate a wide variety of signaling pathways. This broad range of functions makes GPCRs attractive targets for drug discovery. The importance of GPCR research was highlighted in 2012, with the Nobel Prize in chemistry being awarded to Robert Lefkowitz and Brian Kobilka “for studies of G-protein–coupled receptors”.
Based on structure and function, GPCRs are categorized into six classes, A–F. The class A GPCRs, or rhodopsin-like receptors, have been studied extensively due to their association with many types of diseases (2). Within the class A GPCRs is a group that share a highly conserved structural motif (3) and respond to chemokines—small “chemotactic cytokines” that stimulate cell migration, especially that of white blood cells (4). A subfamily of class A GPCRs respond to chemokines that have two cysteine residues near the N-terminus, known as CC chemokines. GPCRs activated by CC chemokines are called CC chemokine receptors or CCRs, and these interactions have been implicated in both pro- and anti-cancer pathways (5).
Cyclin-dependent kinases (CDKs) are promising therapeutic targets in cancer and are currently among the most intensely studied enzymes in drug discovery. The FDA has recently approved three drugs for breast cancer that target members of this kinase subfamily, fueling interest in the entire family. Although broad efforts in drug discovery have produced many CDK inhibitors (CDKIs), few have been characterized in living cells. So just how potent are these compounds in a cellular environment? Are these compounds selective for their intended CDK target, or do they bind many similar kinases in cells? To address these questions, teams at the Structural Genomics Consortium and Promega used the NanoBRET™ Target Engagement technology to uncover surprising patterns of selectivity for touted CDKIs and abandoned clinical leads (1). The results offer exciting opportunities for repurposing some inhibitors as selective chemical probes for lesser-studied CDK family members.
CDKs and CDKIs
Cyclin-dependent kinases (CDKs) regulate a number of key global cellular processes, including cell cycle progression and gene transcription. As the name implies, CDK activity is tightly regulated by interactions with cyclin proteins. In humans, the CDK subfamily consists of 21 members and several are validated drivers of tumorigenesis. For example, CDKs 1, 2, 4 and 6 play a role in cell cycle progression and are validated therapeutic targets in oncology. However, the majority of the remaining CDK family is less studied. For example, some members of the CDK subfamily, such as CDKs 14–18, lack functional annotation and have unclear roles in cell physiology. Others, such as the closely related CDK8/19, are members of multiprotein complexes involved broadly in gene transcription. How these kinases function as members of such large complexes in a cellular context remains unclear, but their activity has been associated with several pathologies, including colorectal cancer. Despite their enormous therapeutic potential, our knowledge of the CDK family members remains incomplete.
The understudied kinome represents a major challenge as well as an exciting opportunity in drug discovery. A team of researchers lead by Nathanael Gray at the Dana Farber Cancer Institute was able to partially elucidate the function of an understudied kinase, Doublecortin-like kinase 1 (DCLK1), in pancreatic ductal adenocarcinoma cells (PDAC). The characterization of DCLK1 in PDAC was realized by developing a highly specific chemical probe (1). Promega NanoBRET™ Target Engagement (TE) technology enabled intracellular characterization of this chemical probe.
The Dark Kinome
Comprised of over 500 proteins, the human kinome is among the broadest class of enzymes in humans and is rife with targets for small molecule therapeutics. Indeed, to date, over 50 small molecule kinase inhibitors have achieved FDA approval for use in treating cancer and inflammatory diseases, with nearly 200 kinase inhibitors in various stages of clinical evaluation (2). Moreover, broad genomic screening efforts have implicated the involvement of a large fraction of kinases in human pathologies (3). Despite such advancements, our knowledge of the kinome is limited to only a fraction of its family members (3,4). For example, currently less than 20% of human kinases are being targeted with drugs in clinical trials. Moreover, only a subset of kinases historically has garnered substantial citations in academic research journals (4). As a result, a large proportion of the human kinome lacks functional annotation; as such, these understudied or “dark” kinases remain elusive to therapeutic intervention (4).
What happens when you engineer a small, super bright luciferase? A generation of new tools. We’ve highlighted many of the papers and new applications that NanoLuc® luciferase has enabled on this blog. Introduced first as a reporter enzyme to assess promoter activity, NanoLuc® has since become the foundation for bioluminescent tools that reveal endogenous protein dynamics, target engagement, protein degradation, immunodetection and more. Much of that biology was largely invisible before these tools existed. So where did NanoLuc® come from, and how does one enzyme power so many research capabilities? Read on for a primer on the technologies and applications built from this enzyme over the last decade.
XWe use cookies and similar technologies to make our website work, run analytics, improve our website, and show you personalized content and advertising. Some of these cookies are essential for our website to work. For others, we won’t set them unless you accept them. To learn more about our approach to Privacy we invite you to Read More
By clicking “Accept All”, you consent to the use of ALL the cookies. However you may visit Cookie Settings to provide a controlled consent.
We use cookies and similar technologies to make our website work, run analytics, improve our website, and show you personalized content and advertising. Some of these cookies are essential for our website to work. For others, we won’t set them unless you accept them. To find out more about cookies and how to manage cookies, read our Cookie Policy.
If you are located in the EEA, the United Kingdom, or Switzerland, you can change your settings at any time by clicking Manage Cookie Consent in the footer of our website.
Necessary cookies are absolutely essential for the website to function properly. These cookies ensure basic functionalities and security features of the website, anonymously.
Cookie
Duration
Description
cookielawinfo-checbox-analytics
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Analytics".
cookielawinfo-checbox-functional
11 months
The cookie is set by GDPR cookie consent to record the user consent for the cookies in the category "Functional".
cookielawinfo-checbox-others
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Other.
cookielawinfo-checkbox-advertisement
1 year
The cookie is set by GDPR cookie consent to record the user consent for the cookies in the category "Advertisement".
cookielawinfo-checkbox-necessary
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookies is used to store the user consent for the cookies in the category "Necessary".
cookielawinfo-checkbox-performance
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Performance".
gdpr_status
6 months 2 days
This cookie is set by the provider Media.net. This cookie is used to check the status whether the user has accepted the cookie consent box. It also helps in not showing the cookie consent box upon re-entry to the website.
lang
This cookie is used to store the language preferences of a user to serve up content in that stored language the next time user visit the website.
viewed_cookie_policy
11 months
The cookie is set by the GDPR Cookie Consent plugin and is used to store whether or not user has consented to the use of cookies. It does not store any personal data.
Analytical cookies are used to understand how visitors interact with the website. These cookies help provide information on metrics the number of visitors, bounce rate, traffic source, etc.
Cookie
Duration
Description
SC_ANALYTICS_GLOBAL_COOKIE
10 years
This cookie is associated with Sitecore content and personalization. This cookie is used to identify the repeat visit from a single user. Sitecore will send a persistent session cookie to the web client.
vuid
2 years
This domain of this cookie is owned by Vimeo. This cookie is used by vimeo to collect tracking information. It sets a unique ID to embed videos to the website.
WMF-Last-Access
1 month 18 hours 24 minutes
This cookie is used to calculate unique devices accessing the website.
_ga
2 years
This cookie is installed by Google Analytics. The cookie is used to calculate visitor, session, campaign data and keep track of site usage for the site's analytics report. The cookies store information anonymously and assign a randomly generated number to identify unique visitors.
_gid
1 day
This cookie is installed by Google Analytics. The cookie is used to store information of how visitors use a website and helps in creating an analytics report of how the website is doing. The data collected including the number visitors, the source where they have come from, and the pages visted in an anonymous form.
Advertisement cookies are used to provide visitors with relevant ads and marketing campaigns. These cookies track visitors across websites and collect information to provide customized ads.
Cookie
Duration
Description
IDE
1 year 24 days
Used by Google DoubleClick and stores information about how the user uses the website and any other advertisement before visiting the website. This is used to present users with ads that are relevant to them according to the user profile.
test_cookie
15 minutes
This cookie is set by doubleclick.net. The purpose of the cookie is to determine if the user's browser supports cookies.
VISITOR_INFO1_LIVE
5 months 27 days
This cookie is set by Youtube. Used to track the information of the embedded YouTube videos on a website.
Performance cookies are used to understand and analyze the key performance indexes of the website which helps in delivering a better user experience for the visitors.
Cookie
Duration
Description
YSC
session
This cookies is set by Youtube and is used to track the views of embedded videos.
_gat_UA-62336821-1
1 minute
This is a pattern type cookie set by Google Analytics, where the pattern element on the name contains the unique identity number of the account or website it relates to. It appears to be a variation of the _gat cookie which is used to limit the amount of data recorded by Google on high traffic volume websites.