Cyclin-dependent kinases (CDKs) are promising therapeutic targets in cancer and are currently among the most intensely studied enzymes in drug discovery. The FDA has recently approved three drugs for breast cancer that target members of this kinase subfamily, fueling interest in the entire family. Although broad efforts in drug discovery have produced many CDK inhibitors (CDKIs), few have been characterized in living cells. So just how potent are these compounds in a cellular environment? Are these compounds selective for their intended CDK target, or do they bind many similar kinases in cells? To address these questions, teams at the Structural Genomics Consortium and Promega used the NanoBRET™ Target Engagement technology to uncover surprising patterns of selectivity for touted CDKIs and abandoned clinical leads (1). The results offer exciting opportunities for repurposing some inhibitors as selective chemical probes for lesser-studied CDK family members.
CDKs and CDKIs
Cyclin-dependent kinases (CDKs) regulate a number of key global cellular processes, including cell cycle progression and gene transcription. As the name implies, CDK activity is tightly regulated by interactions with cyclin proteins. In humans, the CDK subfamily consists of 21 members and several are validated drivers of tumorigenesis. For example, CDKs 1, 2, 4 and 6 play a role in cell cycle progression and are validated therapeutic targets in oncology. However, the majority of the remaining CDK family is less studied. For example, some members of the CDK subfamily, such as CDKs 14–18, lack functional annotation and have unclear roles in cell physiology. Others, such as the closely related CDK8/19, are members of multiprotein complexes involved broadly in gene transcription. How these kinases function as members of such large complexes in a cellular context remains unclear, but their activity has been associated with several pathologies, including colorectal cancer. Despite their enormous therapeutic potential, our knowledge of the CDK family members remains incomplete.
CDK inhibitors (CDKIs) have tremendous therapeutic potential. In the past decade, three CDKIs targeting CDK4/6 have been FDA approved for the treatment of breast cancer, with many other promising leads undergoing clinical trials. Owing to this success with CDK4/6, broad drug discovery efforts have been directed to the other cell cycle CDKs, including CDKs 1, 2 and 7. However, there are multiple issues facing drug discovery efforts directed at CDKs. First, availability of cellular assays that are specific for CDKs has trailed the availability of biochemical CDK assays. Some cell-cycle CDKs, like CDKs 1, 2 and 7, are well characterized enzymatically in a biochemical setting. However, using simple cell-free assays to query inhibitor potency may be insufficient. Although the field is now rife with CDKIs capable of inhibiting enzymatic activity in a biochemical setting, for a CDKI to be useful, it must do more than just inhibit the purified enzyme. Therapeutic efficacy for CDKIs is predicated on their ability to permeate cell membranes, navigate complex intracellular cellular architecture, and physically bind their intended target. A fundamental challenge in developing CDKIs is typified by the question: How will results from cell-free systems that lack this physiological complexity translate into a cellular setting? Since cellular efficacy is dependent on a number of parameters—including compound permeability, selectivity and intrinsic affinity for its intended target—these parameters would be difficult to address in a cell-free system (2). A second challenge in CDK drug development is that the architecture of the CDK active site is highly conserved, so drugs developed for a specific CDK may have unintended collateral affinity for related family members. Such collateral engagement of CDKIs to off-target CDKs can result in unintended side effects in patients. Therefore, determining inhibitor selectivity against the CDK family, in order to rule out collateral engagement of closely related family members, is an important liability to address for drug development programs.
The First Method to Comprehensively Measure CDKI Selectivity in Cells
Although cell-free methods are available for measuring CDK inhibition, querying CDKI potency in cells has been challenging to date. Cellular potency measurements are dependent on the knowledge of intracellular substrates specific to each CDK. Additionally, detection antibodies are needed for measuring specific substrate phosphorylation events from cellular extracts, but these antibodies may not be available. Consequently, only a small fraction of the CDK family can be queried for inhibition in a cellular format.
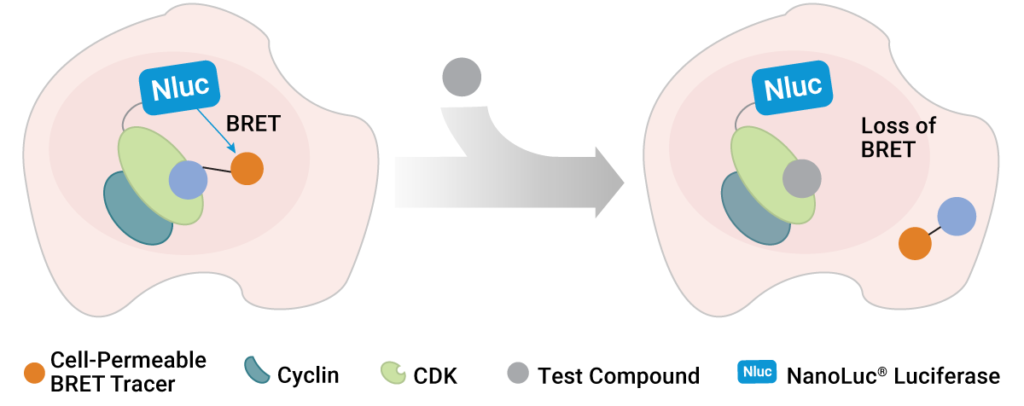
To address this shortcoming, scientists at Promega and the Structural Genomics Consortium developed a method to quantify inhibitor affinity for each CDK in live cells using bioluminescence resonance energy transfer (BRET) (1). Specifically, the team utilized NanoBRET™ Target Engagement technology to develop live-cell assays for all 21 CDKs. In these assays, each CDK is expressed inside living cells with a small, bright NanoLuc® luciferase tag. This assay generates blue light proximal only to the CDK of interest that is fused to the NanoLuc® tag. To detect target engagement, a series of fluorescent BRET tracers were developed and optimized for the entire CDK family. When the tracer binds to the CDK-NanoLuc® fusion, a proximity-dependent BRET signal is generated at that CDK in live cells. Target engagement is then detected by competition with the tracer, commensurate with a loss of BRET. This measurement can be performed in a simple and scalable format, so that many compounds can be tested for affinity and occupancy against many CDKs in a single experiment. Therefore, it became possible for the first time to broadly screen inhibitors for affinity against the entire CDK family in living cells. This allowed the researchers to understand whether the CDKIs are as selective inside cells as advertised in cell-free assays.
Live-Cell CDKI Profiling Reveals Novel Drug Interactions and Opportunities for Drug Repurposing
To evaluate the landscape of CDKI selectivity inside cells, the researchers amassed a broad collection of 46 CDKIs, representing diverse chemical structures, and evaluated target engagement for the entire CDK subfamily (1). In cells, the three FDA approved drugs were proven to be highly selective for their intended targets (CDK 4/6). This result is encouraging since these drugs are being used to treat breast cancer patients. Other CDKIs that are in clinical trials, such as those characterized for CDK 7 and CDK 9, also maintained selectivity for their intended targets when evaluated in live cells. However, the live-cell target engagement results uncovered novel unanticipated interactions for many other compounds that failed clinical trials in the past. This was most pronounced for the CDKIs previously characterized for well-studied family members CDK 1 and CDK 2, which are each involved in cell cycle regulation. Surprisingly, a number of inhibitors previously annotated for CDK 1 and CDK 2 potently engaged understudied members of the family, such as CDK 14–18. Furthermore, three inhibitors were more potent for lesser studied CDKs, such as CDK 8 and CDK 19, despite their annotation for CDK 1 and CDK 2. The engagement potencies were so strong that these CDKIs should be repurposed as selective chemical probes for CDK 8/19. One of the inhibitors (BMS-265246) is an abandoned clinical lead, offering a valuable opportunity to repurpose this failed drug for lesser-studied members of the family.
These findings open the possibility that live-cell profiling will often reveal novel target interactions. For CDKIs and other members of the kinase family, these unintended drug interactions can represent a liability for drug leads. As a result, determining target selectivity in cells can be valuable for de-risking clinical drug candidates. In addition, understanding such interactions may enable new avenues for drug developers to repurpose CDKIs at understudied members. With a comprehensive and simple cell-based method now available, the CDK field is in a stronger position to develop tool compounds and advance clinical candidates for this valuable kinase subfamily.
NanoBRET™ Target Engagement Assays for CDKs and other kinases measure the intracellular affinity of test compounds using a multi-well plate format. Learn More
References
- Wells, C. I., Vasta, J. D. et al. (2020) Quantifying CDK inhibitor selectivity in live cells. Nature Comm. 11, 2743.
- Vasta, J. D. et al. (2018) Quantitative, wide-spectrum kinase profiling in live cells for assessing the effect of cellular ATP on target engagement. Cell Chem Biol 25, 206-214 e211. doi:10.1016/j.chembiol.2017.10.010