Water plays a vital role in countless aspects of daily life—drinking, cooling, recreation and more. However, the same systems that deliver these benefits can also harbor Legionella, a waterborne bacteria responsible for Legionnaires’ disease, a severe form of pneumonia (1). Legionella thrives in stagnant aquatic environments, many of which are human-made and common in modern infrastructure, like in cooling towers, hot tubs and complex building water systems. In this blog, we explore the risks posed by Legionella, the limitations of traditional detection methods and how advanced tools at Promega are transforming water safety monitoring.
Lyophilization is a process designed to remove water from a sample or product through a controlled freezing and vacuum application. The method leverages the triple point of water, where solid, liquid, and gas phases coexist under specific temperature and pressure conditions. The result is a room temperature stable product that is much lighter than the original sample or product.
Imagine a scenario—you’re studying the developmental biology of a species of squid. The squid don’t reproduce in captivity, so females carrying fertilized eggs are collected from the wild and rehomed in your lab’s aquariums. You’ve monitored all the normal aquarium conditions—pH, temperature, salinity—ensuring the animal’s new home mimics its natural environment.
But then, for no reason apparent to you, the clutch of eggs doesn’t develop and doesn’t hatch, derailing your research program until next year when you can collect more adult squid from the wild. What went wrong?
As the SARS‐CoV‐2 pandemic continues to rage across the United States and around the globe, the demand for COVID‐19 testing is increasing. The vast majority of the COVID-19 assays use RT‐qPCR to detect the viral RNA in patient samples such as nasopharyngeal swabs, which are collected and stored in viral or universal transport media (VTM/UTM). The general workflow for these COVID‐19 assays can be broken down as follows:
Collect and store patient samples
Ship samples to testing laboratory
Extract RNA from samples
Amplify and analyze samples
While many companies who manufacture the products that are used in these steps have been able to adapt and significantly increase their production capacities, there are still gaps between the supply of these products and the global test demand. Both the sample collection and storage step and the RNA extraction/purification step have a tendency to bottleneck and experience supply constraints. One way to address these bottlenecks and expand production capacity for these in‐demand products is to evaluate the viability of skipping a step in the workflow, without hindering the ability to detect viral RNA from samples.
The three winners of the 2019 Real-Time PCR Grants have been hard at work in the six months since receiving their grants. Each winner was eligible to receive up to $10,000 in free PCR reagents as well as the opportunity to collaborate with our knowledgeable technical service and training teams.
Abbeah Navasca is a plant pathology researcher with the Tagum Agricultural Development Company, Inc. (TADECO*, Philippines). She is developing treatments for viral infections that affect one of Philippines’ largest and most valuable agricultural exports: bananas. As a result of the qPCR grant, she and two of her colleagues were able to participate in sample preparation and analysis workshops with Promega Technical Services experts in Singapore. During her visit, the team worked through strategies for plant sample preparation and amplified those samples with the GoTaq® 1-Step RT-qPCR System. We had a chance to ask her more before she headed back to her lab.
The three 2019 Real-Time PCR Grant Winners have been hard at work in the six months since winning their grants. Each winner was eligible to receive up to $10,000 in free PCR reagents as well as the opportunity to collaborate with our knowledgeable technical service and training teams.
One of the 2019 winners, Alberto Biscontin (University of Padova, Italy), performs research in the fields of Neurogenetics and Chronobiology. He is looking to shed greater light on the circadian rhythms of the Antarctic krill. Alberto published his most recent analysis in Nature and GoTaq® qPCR Master Mix helped him validate expression of genes for his study.
His qPCR data showed support for internal mechanisms that not only support daily living but also clarified the overwintering process of the krill. Now that Alberto has sized up some zooplankton, we asked him to share a little more about himself and his research:
Q: How long have you been a researcher? A: I have been a researcher since 2012.
Q: How did you decide to research Antarctic krill? A: In 2013, I had the opportunity to join the international Antarctic research program PolarTime. [It] brought together eight research groups with different scientific expertise to study seasonal and daily rhythms in the Antarctic krill Euphausia superba.
Q: When you are not busy at the bench, what do you like to do? A: Traveling. I love strolling through open-air markets.
Q: Are there any tips or tricks you have learned that make your job easier? A: You can easily switch from a classic RT-PCR protocol to a cheaper and faster One-step protocol using the same primers and temperatures.
Q: What comes next? A: I would like to characterize the clock machinery of other polar organisms to understand whether high latitude clocks have developed similar strategies to cope with [the] polar environment. Moreover, a better understanding of marine circadian clocks could help to shed light on the evolution of the animal circadian machinery.
You can find Alberto’s most recent publication in Nature Scientific Reports. The 2020 Real-Time PCR Grant will be coming soon. Be sure to follow us on social media for the most up-to-date information regarding the 2020 Grant, including application deadlines and winner notifications!
PCR amplification with a proofreading polymerase, like Pfu DNA polymerase, will leave you with a blunt end. However, another thermostable DNA polymerase, like Taq DNA Polymerase, adds a single nucleotide base to the 3’ end of the DNA fragment, usually an adenine, creating an “A” overhang. This “A” overhang can create difficulties when cloning the fragment is your end goal. You might consider creating a blunt end with Klenow or adding restriction sites to the ends of your PCR fragment by designing them in your primers. But why go through all those extra steps, when that “A” overhang allows efficient cloning of these fragments into T-Vectors such as the pGEM®-T Vectors? Fewer steps? Who can argue with that?
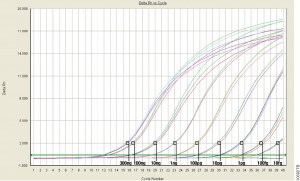
qPCR monitors amplification in real and allows you to measure starting material.
For those of us well versed in traditional, end-point PCR, wrapping our minds and methods around real-time or quantitative (qPCR) can be challenging. Here at Promega Connections, we are beginning a series of blogs designed to explain how qPCR works, things to consider when setting up and performing qPCR experiments, and what to look for in your results.
First, to get our bearings, let’s contrast traditional end-point PCR with qPCR.
End-Point PCR
qPCR
Visualizes by agarose gel the amplified product AFTER it is produced (the end-point)
Visualizes amplification as it happens (in real time) via a detection instrument
Does not precisely measure the starting DNA or RNA
Measures how many copies of DNA or RNA you started with (quantitative = qPCR)
Less expensive; no special instruments required
More expensive; requires special instrumentation
Basic molecular biology technique
Requires slightly more technical prowess
Quantitative PCR (qPCR) can be used to answer the same experimental questions as traditional end-point PCR: Detecting polymorphisms in DNA, amplifying low-abundance sequences for cloning or analysis, pathogen detection and others. However, the ability to observe amplification in real-time and detect the number of copies in the starting material can quantitate gene expression, measure DNA damage, and quantitate viral load in a sample and other applications.
Anytime that you are performing a reaction where something is copied over and over in an exponential fashion, contaminants are just as likely to be copied as the desired input. Quantitative PCR is subject to the same contamination concerns as end-point PCR, but those concerns are magnified because the technique is so sensitive. Avoiding contamination is paramount for generating qPCR results that you can trust.
Use aerosol-resistant pipette tips, and have designated pipettors and tips for pre- and post-amplification steps.
Wear gloves and change them frequently.
Have designated areas for pre- and post-amplification work.
Use reaction “master mixes” to minimize variability. A master mix is a ready-to-use mixture of your reaction components (excluding primers and sample) that you create for multiple reactions. Because you are pipetting larger volumes to make the reaction master mix, and all of your reactions are getting their components from the same master mix, you are reducing variability from reaction to reaction.
Dispense your primers into aliquots to minimize freeze-thaw cycles and the opportunity to introduce contaminants into a primer stock.
These are very basic tips that are common to both end-point and qPCR, but if you get these right, you are off to a good start no matter what your experimental goals are.
If you are looking for more information regarding qPCR, watch this supplementary video below.
Are you looking for more in-depth information about qPCR? Check out our qPCR and RT-qPCR Guide!
PCR is a common technique used in research labs to amplify DNA.
Some thermostable DNA polymerases, including Taq, add a single nucleotide base extension to the 3′ end of amplified DNA fragments. These polymerases usually add an adenine, leaving an “A” overhang. There are several approaches to overcome the cloning difficulties presented by the presence of A overhangs on PCR products. One method involves treating the product with Klenow to create a blunt-ended fragment for subcloning. Another choice is to add restriction sites to the ends of your PCR fragments. You can do this by incorporating the desired restriction sites into the PCR primers. After amplification, the PCR product is digested and subcloned into the cloning vector. Take care when using this method, as not all restriction enzymes efficiently cleave at the ends of DNA fragments, and you may not be able to use every restriction enzyme you desire. There is some useful information about cutting with restriction sites close to the end of linear fragments in the Restriction Enzyme Resource Guide. Also, some restriction enzymes require extra bases outside the recognition site, adding further expense to the PCR primers as well as risk of priming to unrelated sequences in the genome.
Have you ever thought about plant viruses? Unless you’re a farmer or avid gardener, probably not. And yet, for many people the battle against agricultural viruses never ends. Plant viruses cause billions of dollars in damage every year and leave millions of people food insecure (1–2), making viruses a major barrier to meeting the United Nations’ global sustainable development goal of Zero Hunger by 2030.
At the University of Western Australia, Senior Research Fellow Dr. Laura Boykin is using genomics and supercomputing to tackle the problem of viral plant diseases. In a recent study, Dr. Boykin and her colleagues used genome sequencing to inform disease management in cassava crops. For this work, they used the MinION, a miniature, portable sequencer made by Oxford Nanopore Technologies, to fully sequence the genomes of viruses infecting cassava plants.
Cassava (Manihot esculenta) is one of the 5 most important calorie sources worldwide (3). Over 800 million people rely on cassava for food and/or income (4). Cassava is susceptible to a group of viruses called begomoviruses, which are transmitted by whiteflies. Resistant cassava varieties are available. However, these resistant plants are usually only protected against a small number of begomoviruses, so proper deployment of these plants means farmers must know both whether their plants are infected and, if so, the strain of virus that’s causing the infection.
XWe use cookies and similar technologies to make our website work, run analytics, improve our website, and show you personalized content and advertising. Some of these cookies are essential for our website to work. For others, we won’t set them unless you accept them. To learn more about our approach to Privacy we invite you to Read More
By clicking “Accept All”, you consent to the use of ALL the cookies. However you may visit Cookie Settings to provide a controlled consent.
We use cookies and similar technologies to make our website work, run analytics, improve our website, and show you personalized content and advertising. Some of these cookies are essential for our website to work. For others, we won’t set them unless you accept them. To find out more about cookies and how to manage cookies, read our Cookie Policy.
If you are located in the EEA, the United Kingdom, or Switzerland, you can change your settings at any time by clicking Manage Cookie Consent in the footer of our website.
Necessary cookies are absolutely essential for the website to function properly. These cookies ensure basic functionalities and security features of the website, anonymously.
Cookie
Duration
Description
cookielawinfo-checbox-analytics
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Analytics".
cookielawinfo-checbox-functional
11 months
The cookie is set by GDPR cookie consent to record the user consent for the cookies in the category "Functional".
cookielawinfo-checbox-others
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Other.
cookielawinfo-checkbox-advertisement
1 year
The cookie is set by GDPR cookie consent to record the user consent for the cookies in the category "Advertisement".
cookielawinfo-checkbox-necessary
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookies is used to store the user consent for the cookies in the category "Necessary".
cookielawinfo-checkbox-performance
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Performance".
gdpr_status
6 months 2 days
This cookie is set by the provider Media.net. This cookie is used to check the status whether the user has accepted the cookie consent box. It also helps in not showing the cookie consent box upon re-entry to the website.
lang
This cookie is used to store the language preferences of a user to serve up content in that stored language the next time user visit the website.
viewed_cookie_policy
11 months
The cookie is set by the GDPR Cookie Consent plugin and is used to store whether or not user has consented to the use of cookies. It does not store any personal data.
Analytical cookies are used to understand how visitors interact with the website. These cookies help provide information on metrics the number of visitors, bounce rate, traffic source, etc.
Cookie
Duration
Description
SC_ANALYTICS_GLOBAL_COOKIE
10 years
This cookie is associated with Sitecore content and personalization. This cookie is used to identify the repeat visit from a single user. Sitecore will send a persistent session cookie to the web client.
vuid
2 years
This domain of this cookie is owned by Vimeo. This cookie is used by vimeo to collect tracking information. It sets a unique ID to embed videos to the website.
WMF-Last-Access
1 month 18 hours 24 minutes
This cookie is used to calculate unique devices accessing the website.
_ga
2 years
This cookie is installed by Google Analytics. The cookie is used to calculate visitor, session, campaign data and keep track of site usage for the site's analytics report. The cookies store information anonymously and assign a randomly generated number to identify unique visitors.
_gid
1 day
This cookie is installed by Google Analytics. The cookie is used to store information of how visitors use a website and helps in creating an analytics report of how the website is doing. The data collected including the number visitors, the source where they have come from, and the pages visted in an anonymous form.
Advertisement cookies are used to provide visitors with relevant ads and marketing campaigns. These cookies track visitors across websites and collect information to provide customized ads.
Cookie
Duration
Description
IDE
1 year 24 days
Used by Google DoubleClick and stores information about how the user uses the website and any other advertisement before visiting the website. This is used to present users with ads that are relevant to them according to the user profile.
test_cookie
15 minutes
This cookie is set by doubleclick.net. The purpose of the cookie is to determine if the user's browser supports cookies.
VISITOR_INFO1_LIVE
5 months 27 days
This cookie is set by Youtube. Used to track the information of the embedded YouTube videos on a website.
Performance cookies are used to understand and analyze the key performance indexes of the website which helps in delivering a better user experience for the visitors.
Cookie
Duration
Description
YSC
session
This cookies is set by Youtube and is used to track the views of embedded videos.
_gat_UA-62336821-1
1 minute
This is a pattern type cookie set by Google Analytics, where the pattern element on the name contains the unique identity number of the account or website it relates to. It appears to be a variation of the _gat cookie which is used to limit the amount of data recorded by Google on high traffic volume websites.