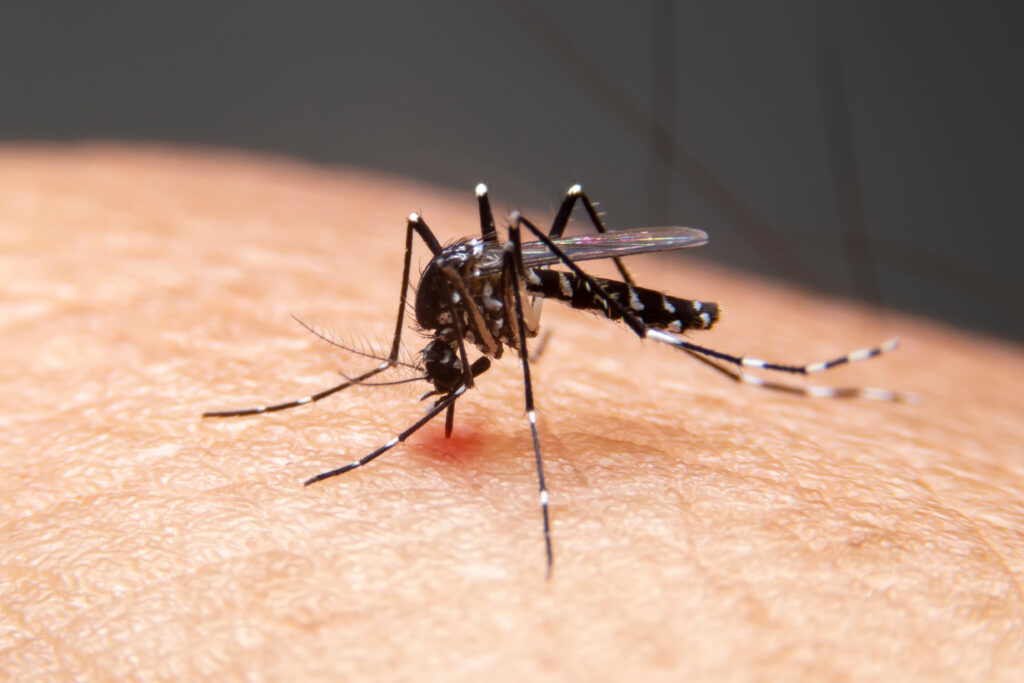
Festival season is here—and apparently, mosquitoes got tickets too.
If you have ever been the person in your friend group who ends a summer concert covered in large, itchy welts while everyone else goes home bite-free, you are not imagining things. Some people really are mosquito magnets.
A new study, aptly titled “Blood, Sweat, and Beers,” set out to uncover what makes certain humans irresistible to mosquitoes. But instead of a sterile lab or a rainforest expedition, this experiment took place at one of the Netherlands’ biggest music festivals; Lowlands, a three-day party with 65,000 attendees, questionable hygiene and plenty of beer. In other words: the perfect breeding ground for this science experiment.
Transfection is a core technique in molecular biology used to introduce foreign nucleic acids—such as DNA, RNA, or small RNAs like siRNA, shRNA, and miRNA—into eukaryotic cells. This enables researchers to manipulate gene expression and study cellular processes, disease mechanisms and therapeutic strategies (1).
Advances in transfection technology now support a range of nucleic acid types and cell models. Researchers can pursue transient or stable expression to achieve specific goals: knocking down transcripts, expressing proteins, or probing promoter activity in systems from immortalized lines to stem cells (1).
RT-qPCR (reverse transcription quantitative PCR) is a powerful technique for quantifying RNA expression—but it doesn’t always cooperate. Even when you’ve followed the protocol carefully, unexpected results can appear: flat curves, unexpected Cq values, or inconsistent replicates. When that happens, you’re left wondering… what went wrong?
In this blog, we’ll walk through five key questions to help you troubleshoot RT-qPCR issues with confidence. From common errors to more stubborn challenges, we’ll also explore what to consider when technique isn’t fully the problem—and when it might be time to rethink your reagents.
Today’s blog is written by guest blogger, Gabriela Saldanha, Senior Product Marketing Manager at Promega.
Quantitative PCR (qPCR) is an indispensable tool for nucleic acid analysis, widely used in research, clinical diagnostics and applied sciences. Its sensitivity and specificity make it a powerful method for detecting and quantifying DNA and RNA targets. However, qPCR reactions are highly susceptible to inhibitors—substances that interfere with enzyme activity, primer binding, or fluorescent signal detection. These inhibitors can originate from biological samples, environmental contaminants, or laboratory reagents, potentially leading to inaccurate quantification, poor amplification efficiency, or complete reaction failure.
Today’s blog is written by guest blogger, Sameer Moorji, Director, Applied Markets.
People’s diets are frequently influenced by a wide range of variables; with environment, socioeconomic status, religion, and culture being a few of the key influencers. The Muslim community serves as one illustration of how culture and religion can hold influence over people’s eating habits.
Muslims, who adhere to Islamic teachings derived from the Qur’an, frequently base dietary choices on a food’s halal status, whether it is permissible to consume, or haram status, forbidden to consume. With the population of Muslims expected to expand from 1.6 billion in 2010 to 2.2 billion by 2030, the demand for halal products is anticipated to surge (2).
By 2030, the global halal meat market is projected to reach over $300 billion dollars, with Asia-Pacific and the Middle East regions being the largest consumers and producers of halal meat products (3). Furthermore, increasing awareness and popularity of halal meat among non-Muslim consumers, as well as strengthening preference for ethical and high-quality meat, are all contributing to demand.
RNA polymerase unwinds DNA strands for transcription.
Transcription is the production of RNA from a DNA sequence. It’s a necessary life process in most cells. Transcription performed in vitro is also a valuable technique for research applications—from gene expression studies to the development of RNA virus vaccines.
During transcription, the DNA sequence is read by RNA polymerase to produce a complimentary, antiparallel RNA strand. This RNA strand is called a primary transcript, often referred to as an RNA transcript. In vitro transcription is a convenient method for generating RNA in a controlled environment outside of a cell.
In vitro transcription offers flexibility when choosing a DNA template, with a few requirements. The template must be purified, linear, and include a double stranded promoter region. Acceptable template types are plasmids or cloning vectors, PCR products, synthetic oligos (oligonucleotides), and cDNA (complimentary DNA).
In vitro transcription is used for production of large amounts of RNA transcripts for use in many applications including gene expression studies, RNA interference studies (RNAi), generation of guide RNA (gRNA) for use in CRISPR, creation of RNA standards for quantification of results in reverse-transcription quantitative PCR (RT-qPCR), studies of RNA structure and function, labeling of RNA probes for blotting and hybridization or for RNA:protein interaction studies, and preparation of specific cDNA libraries, just to name a few!
In vitro transcription can also be applied in general virology to study the effects of an RNA virus on a cell or an organism, and in development and production of RNA therapeutics and RNA virus vaccines. The large quantity of viral RNA produced through in vitro transcription can be used as inoculation material for viral infection studies. Viral mRNA transcripts, typically coding for a disease-specific antigen, can be quickly created through in vitro transcription, and used in the production of vaccines and therapeutics.
Traditionally, scientists have relied on flat,
two-dimensional cell cultures grown on substrates such as tissue culture
polystyrene (TCPS) to study cellular physiology. These models are simple and
cost-effective to culture and process. Within the last decade, however, three-dimensional
(3D) cell cultures have become increasingly popular because they are more
physiologically relevant and better represent in vivo conditions.
Have you ever thought about plant viruses? Unless you’re a farmer or avid gardener, probably not. And yet, for many people the battle against agricultural viruses never ends. Plant viruses cause billions of dollars in damage every year and leave millions of people food insecure (1–2), making viruses a major barrier to meeting the United Nations’ global sustainable development goal of Zero Hunger by 2030.
At the University of Western Australia, Senior Research Fellow Dr. Laura Boykin is using genomics and supercomputing to tackle the problem of viral plant diseases. In a recent study, Dr. Boykin and her colleagues used genome sequencing to inform disease management in cassava crops. For this work, they used the MinION, a miniature, portable sequencer made by Oxford Nanopore Technologies, to fully sequence the genomes of viruses infecting cassava plants.
Cassava (Manihot esculenta) is one of the 5 most important calorie sources worldwide (3). Over 800 million people rely on cassava for food and/or income (4). Cassava is susceptible to a group of viruses called begomoviruses, which are transmitted by whiteflies. Resistant cassava varieties are available. However, these resistant plants are usually only protected against a small number of begomoviruses, so proper deployment of these plants means farmers must know both whether their plants are infected and, if so, the strain of virus that’s causing the infection.
In general, people like to know that their food is what the label says it is. It’s a real bummer to find out that beef lasagna you just ate was actually horsemeat. Plus, there are many religious, ethical and medical reasons to be cognizant of what you eat. Someone who’s gluten intolerant and Halal probably doesn’t want a bite of that BLT.
Labels don’t always accurately reflect what is in food. So how do we confirm that we are in fact buying crab, and not whitefish with a side of Vibrio contamination?
For the most part, it comes down to separation science. Scientists and technicians use various chromatographic methods, such as gas chromatography, liquid chromatography, and mass spectrometry, to separate the complex mixture of molecules in food into individual components. By first mapping out the molecular profile of reference samples, they can then take an unknown sample and compare its profile to what it should look like. If the two don’t match up, an analyst would assume that the unknown is not what it claims to be. Continue reading “Of Mice and Microbes: The Science Behind Food Analysis”
Implementing automated nucleic acid purification or making changes to your high-throughput (HT) workflow can be complicated and time-consuming. There are also many barriers to success such as challenging samples types and maintaining desirable downstream results that can add to the stress, not to mention actually getting the robotic instrumentation to do what you want it to. All of this makes it easy to understand why many labs avoid automating or own expensive instrumentation that goes unused.
XWe use cookies and similar technologies to make our website work, run analytics, improve our website, and show you personalized content and advertising. Some of these cookies are essential for our website to work. For others, we won’t set them unless you accept them. To learn more about our approach to Privacy we invite you to Read More
By clicking “Accept All”, you consent to the use of ALL the cookies. However you may visit Cookie Settings to provide a controlled consent.
We use cookies and similar technologies to make our website work, run analytics, improve our website, and show you personalized content and advertising. Some of these cookies are essential for our website to work. For others, we won’t set them unless you accept them. To find out more about cookies and how to manage cookies, read our Cookie Policy.
If you are located in the EEA, the United Kingdom, or Switzerland, you can change your settings at any time by clicking Manage Cookie Consent in the footer of our website.
Necessary cookies are absolutely essential for the website to function properly. These cookies ensure basic functionalities and security features of the website, anonymously.
Cookie
Duration
Description
cookielawinfo-checbox-analytics
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Analytics".
cookielawinfo-checbox-functional
11 months
The cookie is set by GDPR cookie consent to record the user consent for the cookies in the category "Functional".
cookielawinfo-checbox-others
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Other.
cookielawinfo-checkbox-advertisement
1 year
The cookie is set by GDPR cookie consent to record the user consent for the cookies in the category "Advertisement".
cookielawinfo-checkbox-necessary
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookies is used to store the user consent for the cookies in the category "Necessary".
cookielawinfo-checkbox-performance
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Performance".
gdpr_status
6 months 2 days
This cookie is set by the provider Media.net. This cookie is used to check the status whether the user has accepted the cookie consent box. It also helps in not showing the cookie consent box upon re-entry to the website.
lang
This cookie is used to store the language preferences of a user to serve up content in that stored language the next time user visit the website.
viewed_cookie_policy
11 months
The cookie is set by the GDPR Cookie Consent plugin and is used to store whether or not user has consented to the use of cookies. It does not store any personal data.
Analytical cookies are used to understand how visitors interact with the website. These cookies help provide information on metrics the number of visitors, bounce rate, traffic source, etc.
Cookie
Duration
Description
SC_ANALYTICS_GLOBAL_COOKIE
10 years
This cookie is associated with Sitecore content and personalization. This cookie is used to identify the repeat visit from a single user. Sitecore will send a persistent session cookie to the web client.
vuid
2 years
This domain of this cookie is owned by Vimeo. This cookie is used by vimeo to collect tracking information. It sets a unique ID to embed videos to the website.
WMF-Last-Access
1 month 18 hours 24 minutes
This cookie is used to calculate unique devices accessing the website.
_ga
2 years
This cookie is installed by Google Analytics. The cookie is used to calculate visitor, session, campaign data and keep track of site usage for the site's analytics report. The cookies store information anonymously and assign a randomly generated number to identify unique visitors.
_gid
1 day
This cookie is installed by Google Analytics. The cookie is used to store information of how visitors use a website and helps in creating an analytics report of how the website is doing. The data collected including the number visitors, the source where they have come from, and the pages visted in an anonymous form.
Advertisement cookies are used to provide visitors with relevant ads and marketing campaigns. These cookies track visitors across websites and collect information to provide customized ads.
Cookie
Duration
Description
IDE
1 year 24 days
Used by Google DoubleClick and stores information about how the user uses the website and any other advertisement before visiting the website. This is used to present users with ads that are relevant to them according to the user profile.
test_cookie
15 minutes
This cookie is set by doubleclick.net. The purpose of the cookie is to determine if the user's browser supports cookies.
VISITOR_INFO1_LIVE
5 months 27 days
This cookie is set by Youtube. Used to track the information of the embedded YouTube videos on a website.
Performance cookies are used to understand and analyze the key performance indexes of the website which helps in delivering a better user experience for the visitors.
Cookie
Duration
Description
YSC
session
This cookies is set by Youtube and is used to track the views of embedded videos.
_gat_UA-62336821-1
1 minute
This is a pattern type cookie set by Google Analytics, where the pattern element on the name contains the unique identity number of the account or website it relates to. It appears to be a variation of the _gat cookie which is used to limit the amount of data recorded by Google on high traffic volume websites.