In 1963, Jennifer Harvey was studying Moloney murine leukemia virus (MMLV) at the cancer research department of the London Hospital Research Laboratories. After routine transfers of plasma from MMLV-infected rats to mice, she made an unusual discovery. In addition to the expected leukemia, the mice that received the plasma developed solid tumors (soft-tissue sarcomas), primarily in the spleen (1). A few years later, Werner Kirsten at the University of Chicago observed similar results working with mouse erythroblastosis virus (MEV) (2).
Subsequent research, with the advent of genome sequencing, showed that a cellular rat gene had been incorporated into the viral genome in both cases (3). These genomic sequences contained a mutation later shown to be responsible for the development of sarcomas, and the word “oncogene” became a common part of the vocabulary in cancer publications during the early 1980s (4). Harvey’s discovery led to the naming of the corresponding rat sarcoma oncogene as HRAS, while Kirsten’s related oncogene was named KRAS. Several laboratories, working independently, cloned the human homolog of the viral HRAS gene in 1982 (3). The human KRAS gene was cloned shortly thereafter, as well as a third RAS gene, named NRAS (3). Additional studies showed that a single point mutation in each of these genes led to oncogenic activation, and they have been popular targets for anticancer drug discovery efforts ever since.
All in the Family
KRAS, HRAS and NRAS are all small GTPases (20–25kDa): proteins that bind and hydrolyze guanosine triphosphate (GTP) to guanosine diphosphate (GDP). GTP binding activates RAS, altering the protein conformation; activated RAS can then bind to a downstream effector protein. GTP hydrolysis leads to release of the effector protein, a phosphate ion and inactive RAS. Thus, the signal is transmitted along the cascade, with the GTP-GDP conversion reaction acting like an on-off switch. Currently, there are over 150 known members of the RAS superfamily of proteins (3).
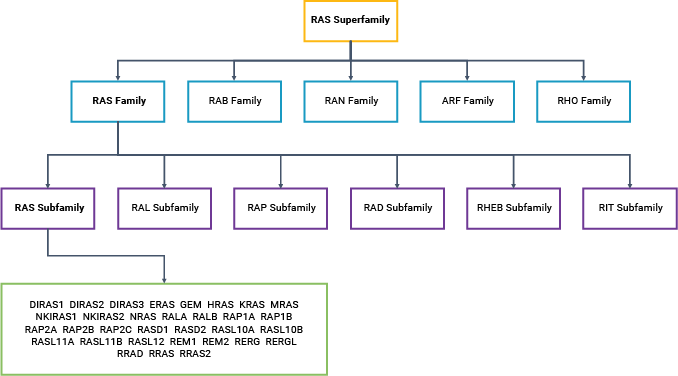
Typically, RAS signaling starts at the cell membrane with the activation of a receptor, such as receptor tyrosine kinases (RTKs), G-protein coupled receptors (GPCRs), or an integrin family member (6). This widely studied signaling pathway is triggered by the binding of a growth factor, such as epidermal growth factor (EGF), to its RTK receptor at the cell surface. This event sets off a cascade that includes sequential activation of RAS, RAF, mitogen-activated protein/extracellular signal-regulated kinase (MEK) and extracellular signal-regulated kinase (ERK). The MAPK-ERK pathway, also known as the RAS-RAF-MEK-ERK pathway, controls a range of cellular events—most importantly, cell cycle entry, differentiation and proliferation. Thus, it is not surprising that dysregulation of the MAPK-ERK pathway is the most common cause of cancer in humans. RAS subfamily mutations are associated with a wide range of cancer types, including melanomas and leukemias, as well as pancreatic, colorectal and thyroid cancers (7, 8). Within the RAS subfamily, KRAS is the most frequently mutated member (9).
Targeting the RAS Pathway
Initial RAS-targeted drug discovery efforts that directly targeted the proteins proved challenging, largely due to the protein structure, and RAS was long considered an “undruggable” protein (10). Therefore, recent RAS-targeted drug discovery strategies have focused on downstream components of the MAPK pathway, particularly BRAF and MEK; however, drug resistance is a common occurrence with the use of these inhibitors (11, 12).
Research targeting the KRAS-G12C mutant has shown promise, with a covalent inhibitor (AMG 510 or Lumakras) receiving accelerated FDA approval for treatment of KRAS-G12C-mutated locally advanced or metastatic non-small-cell lung cancer. Other small-molecule drugs that are direct, allele-specific inhibitors of mutant RAS proteins are proving their effectiveness, either alone or in combination (reviewed in 13).
The challenges associated with targeting specific RAS mutants using small-molecule inhibitors have led to consideration of alternative strategies. Foremost among these is targeted protein degradation (TPD). Instead of inhibiting the target protein, this method tags it for destruction using the cell’s own “garbage disposal” system. A proof-of-concept study used both a KRAS-specific degrader and an intracellular single-domain antibody pan-RAS degrader—active against a range of RAS mutants—illustrating the potential of TPD (14).
Further understanding of the complex signaling pathways involved in the development of cancer will spur the discovery and development of new and more effective anticancer therapies. At the same time, oncology research can benefit from advances in computational modeling methods, such as the “digital twins” concept (15). This concept aims to create a digital representation, or computational avatar, of each patient so that potential therapies can be tested in silico before being administered in the clinic. Such approaches will help to personalize therapies and maximize effectiveness for each patient.
Learn more about products and technologies to facilitate RAS-targeted drug discovery with our applications page.
References
- Harvey, J.J. (1964) An unidentified virus which causes the rapid production of tumors in mice. Nature 204, 1104–1105.
- Kirsten, W.H. and Mayer, L.A. (1967) Morphologic responses to a murine erythroblastosis virus. J. Natl. Cancer Inst. 39, 311–335.
- Fernández-Medarde, A. et al. (2021) 40 years of RAS—a historic overview. Genes 12, 681.
- Tsuchia, N. et al. (2016) Kirsten Ras oncogene: Significance of its discovery in human cancer research. Oncotarget 7(29), 46717–46733.
- Liu, F. et al. (2018) Targeting ERK, an Achilles’ heel of the MAPK pathway, in cancer therapy. Acta Pharm. Sin. B 8(4), 552–562.
- Gimple, R.C. and Wang, X. (2019) RAS: Striking at the core of the oncogenic circuitry. Front. Oncol. 9, 965.
- Cerami, E. et al. (2012) The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404.
- Gao, J. et al. (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science Signal. 6, pl1.
- Hobbs, G.A. et al. (2016) RAS isoforms and mutations in cancer at a glance. J Cell Sci. 129, 1287–1292.
- Fernández-Medarde A. and Santos, E. (2011) Ras in cancer and developmental diseases. Genes Cancer 2, 344–358.
- Little, A.S. et al. (2013) Mechanisms of acquired resistance to ERK1/2 pathway inhibitors. Oncogene 32, 1207–1215.
- Moriceau, G. et al. (2015) Tunable-combinatorial mechanisms of acquired resistance limit the efficacy of BRAF/MEK cotargeting but result in melanoma drug addiction. Cancer Cell 27, 240–256.
- Moore, A. R. et al. (2020) RAS-targeted therapies: is the undruggable drugged? Nat. Rev. Drug Discov. 19(8), 533–552.
- Bery, N. et al. (2020) A potent KRAS macromolecule degrader specifically targeting tumours with mutant KRAS. Nat. Comm. 11, 3233.
- Lee, S. et al. (2020) Targeting MAPK signaling in cancer: mechanisms of drug resistance and sensitivity. Int. J. Mol. Sci. 21, 1102.
Related Posts
Latest posts by Ken Doyle (see all)
- Will Artificial Intelligence (AI) Transform the Future of Life Science Research? - February 1, 2024
- RAF Inhibitors: Quantifying Drug-Target Occupancy at Active RAS-RAF Complexes in Live Cells - September 5, 2023
- Synthetic Biology: Minimal Cell, Maximal Opportunity - July 25, 2023