
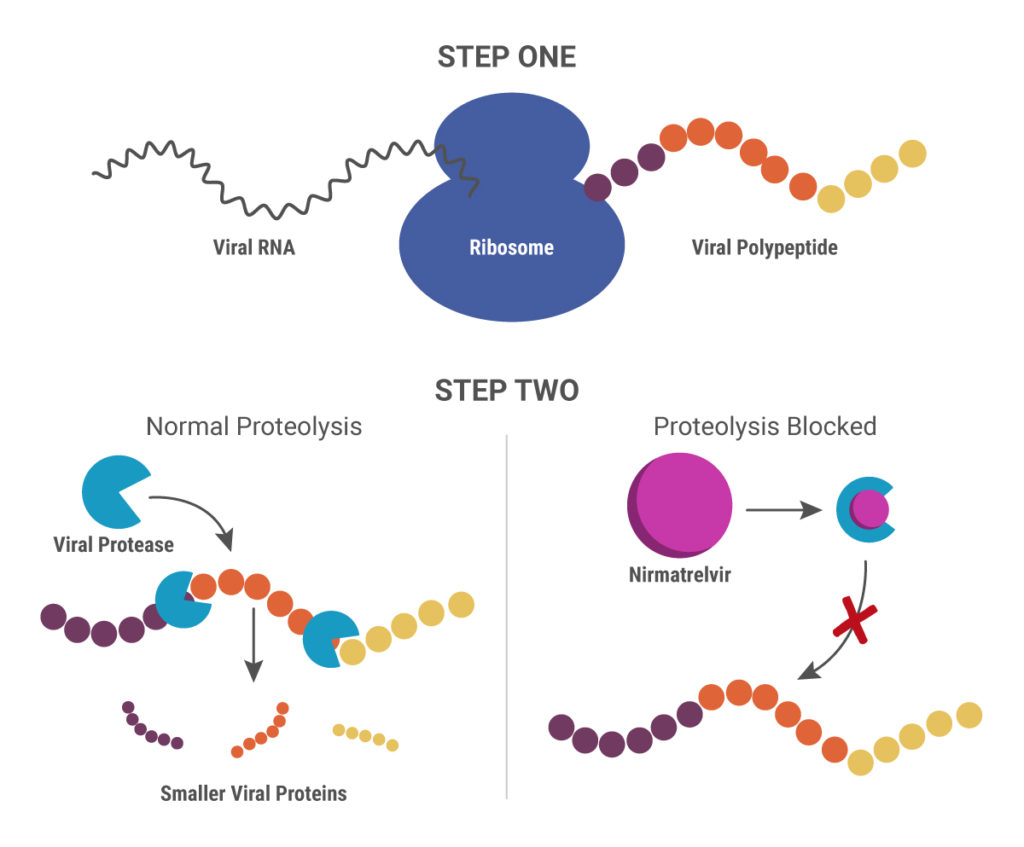
Cytochrome P450 (CYP) inhibitors are often used as boosting agents in combination with other drugs. This drug development strategy is front and center for Paxlovid, the new anti-SARS-CoV-2 treatment from Pfizer. Paxlovid is a combination therapy, comprised of two protease inhibitors, nirmatrelvir and ritonavir. It significantly reduces the risk of COVID-19 hospitalization in high-risk adults and is ingested orally rather than injected, which is an advantage over other SARS-CoV-2 treatments, such as Remdesivir.
Nirmatrelvir was originally developed by Pfizer almost 20 years ago to treat HIV and works by blocking enzymes that help viruses replicate. Pfizer created another version of this drug to combat SARS in 2003, but, once that outbreak ended, further development was put on pause until the advent of the COVID-19 pandemic. After developing an intravenous form of nirmatrelvir early in the pandemic, Pfizer created another version that can be taken orally and combined it with ritonavir.
When ritonavir was originally developed, it wasn’t considered particularly useful because it metabolized so quickly in the body. Now it is recognized as a pharmacokinetic enhancer in combination with other drugs. Ritonivir inhibits CYP3A4, an enzyme which plays a key role in the metabolism of drugs and xenobiotics. By inhibiting CYP3A4, ritonivir slows the metabolism of other drugs. In the case of Paxlovid, this allows nirmatrelvir to stay in the body longer at a high enough concentration to be effective against the virus. This ultimately means that patients can be given lower doses of the drug with reducing efficacy.



![Fig 4. Four point MMOA screen for tideglusib and GW8510. Time dependent inhibition was evaluated by preincubation of TbGSK3β with 60 nM tideglusib and 6 nM GW-8510 with 10μM and 100μM ATP. (A). Tideglusib [60 nM] in 10μM ATP. (B). GW8510 [60 nM] in 10μM ATP. (C.) Tideglusib [60 nM] at 100μM ATP. (D.) GW8510 [60 nM] at 100μM ATP. All reactions preincubated or not preincubated with TbGSK3β for 30 min at room temperature. Experiments run with 10μM GSM peptide, 10μM ATP, and buffer. Minute preincubation (30 min) was preincubated with inhibitor, TbGSK3β, GSM peptide, and buffer. ATP was mixed to initiate reaction. No preincubation contained inhibitor, GSM peptide, ATP, and buffer. The reaction was initiated with TbGSK3β. Reactions were run at room temperature for 5 min and stopped at 80°C. ADP formed was measured by ADP-Glo kit. Values are mean +/- standard error. N = 3 for each experiment and experiments were run in duplicates. Control reactions contained DMSO and background was determined using a zero time incubation and subtracted from all reactions. Black = 30 min preincubation Grey = No preincubation.](https://www.promegaconnections.com/wp-content/uploads/2016/04/journal.pntd_.0004506.g004-243x300.png)