Protein phosphorylation is a very important protein post-translational modification that controls many cellular processes including metabolism, transcriptional and translation regulation, degradation of proteins, cellular signaling and communication, proliferation, differentiation, and cell survival (1). Approximately 35% of human proteins are phosphorylated. Phosphoproteins are low in abundance, and, therefore, are challenging to detect and characterize by mass spectrometry. Different enrichment systems have been developed to isolate phosphopeptides. Among these techniques, immobilized metal affinity chromatography (IMAC) using Fe3+ and Ga3+ has been widely used for the enrichment of phosphopeptides.
Protein phosphorylation is a very important protein post-translational modification that controls many cellular processes including metabolism, transcriptional and translation regulation, degradation of proteins, cellular signaling and communication, proliferation, differentiation, and cell survival (1). Approximately 35% of human proteins are phosphorylated. Phosphoproteins are low in abundance, and, therefore, are challenging to detect and characterize by mass spectrometry. Different enrichment systems have been developed to isolate phosphopeptides. Among these techniques, immobilized metal affinity chromatography (IMAC) using Fe3+ and Ga3+ has been widely used for the enrichment of phosphopeptides.
Typical experimental workflows are tedious and consist of numerous steps, including sample collection and cell lysis. One of the major challenges of the process is to maintain the in vivo phosphorylation state of the proteins throughout the preparation process
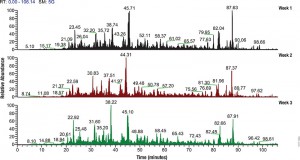
To evaluate the effect of sample collection protocols on the global phosphorylation status of the cell, a recent paper by Kashin et al. compared different sample workflows by metabolic labeling and quantitative mass spectrometry on Saccharomyces cerevisiae cell cultures (2).
Three different sample collection workflows were evaluated: two that used denaturating conditions and involved mixing of cell cultures with an excess of either ethanol (EtOH) at −80 °C or trichloroacetic acid (TCA), and a third under nondenaturing conditions and washing the cells in PBS.
Their data suggest that either TCA or EtOH sample collection protocols introduced lower collection bias than the PBS protocol. It was also suggested that similar studies be carried out to determine what effects sample preparation has on other post translation modifications such as acetylation or ubiquitination.
Literature Cited
- Thingholm T.E. et al, (2009) Analytical strategies for phosphoproteomics. Proteomics 9,1451–68
- Kanshin, E. et al. (2015) Sample Collection Method Bias Effects in Quantitative Phosphoproteomics. J Proteome Res. 14, 2998-04.
 Filter-aided sample preparation (FASP) method is used for the on-filter digestion of proteins prior to mass-spectrometry-based analyses (1,2). FASP was designed for the removal of detergents, and chaotropes that were used for sample preparation. In addition, FASP removes components such as salts, nucleic acids and lipids. Akylation of reduced cysteine residues is also carried out on filter, after which protein is proteolyzed by use of trypsin on filter in the optimal buffer of the enzyme. Subsequent elution and desalting of the peptide-rich solution then provides a sample ready for LC–MS/MS analysis.
Filter-aided sample preparation (FASP) method is used for the on-filter digestion of proteins prior to mass-spectrometry-based analyses (1,2). FASP was designed for the removal of detergents, and chaotropes that were used for sample preparation. In addition, FASP removes components such as salts, nucleic acids and lipids. Akylation of reduced cysteine residues is also carried out on filter, after which protein is proteolyzed by use of trypsin on filter in the optimal buffer of the enzyme. Subsequent elution and desalting of the peptide-rich solution then provides a sample ready for LC–MS/MS analysis.