Studying proteins in their native biological context has long been a major challenge in molecular biology. Traditional methods, although widely used, often distort the actual cellular environment and limit functional interpretation. Techniques like antibody-based detection or plasmid-driven overexpression can introduce artifacts and do not allow real-time analysis in living cells.
In this context, the need for tools that enable the observation of proteins as they naturally occur, under physiological conditions, and within live cells is becoming increasingly evident in molecular biology.
Studying protein function in live cells is limited by the tools available to analyze the expression and interactions of those proteins. Although mass spectrometry and antibody-based protein detection are valuable technologies for protein analysis, both methods have drawbacks that limit the range of targets and contexts in which proteins can be investigated.
Mass spectrometry is often poor at detecting low-abundance proteins. Antibody-based techniques require high quality, specific antibodies, which can be difficult to impossible to acquire. Both methods require cell lysis, preventing real-time analysis and limiting the physiological relevance, and both methods can be limiting for higher-throughput analysis. While plasmid-based overexpression of tagged target proteins simplifies detection and can allow for real time analysis, protein levels don’t typically resemble endogenous levels. Overexpression also has the potential to create experimental artifacts or limit the dynamic range of an observed response.
In 2018, Promega R&D scientists published a paper in ACS Chemical Biology demonstrating the use of CRISPR/Cas9 to integrate the 11 amino acid, bioluminescent HiBiT tag directly into the genome to serve as an easily measured reporter for endogenous proteins. This provides a highly quantitative method for investigating cellular protein dynamics that sidesteps the need for cloning and other drawbacks to conventional methods, including the ability to measure changing protein dynamics in real-time. (For more details about CRISPR/Cas9 knock-in tagging and other applications, read this blog.)
While their findings showed that this method provides efficient and specific tagging of endogenous proteins, the research was limited to just five different proteins within a single signaling pathway in two cell lines. This left unanswered questions about whether this approach was scalable, had broader applications and how accurately the natural biology of the cells was represented.
You have identified and cloned your protein of interest, but you want to explore its function. A protein fusion tag might help with your investigation. However, choosing a tag for your protein depends on what experiments you are planning. Do you want to purify the protein? Would you like to identify interacting proteins by performing pull-down assays? Are you interested in examining the endogenous biology of the protein? Here we cover the advantages and disadvantages of some protein tags to help you select the one that best suits your needs.
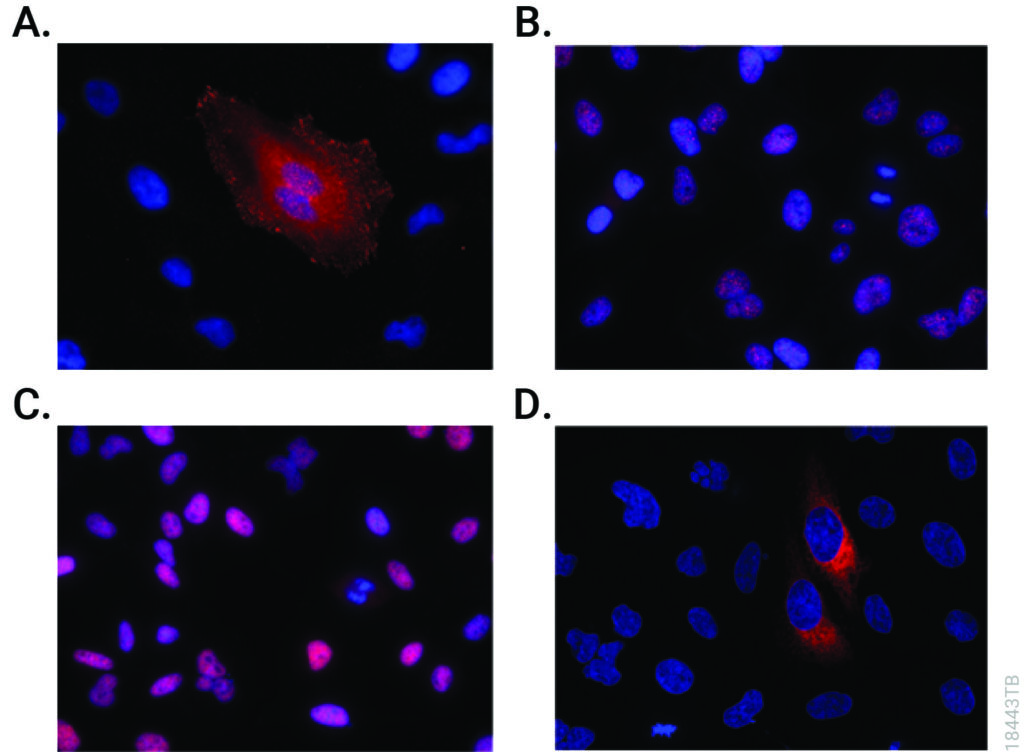
CRISPR-Cas9 editing knocked-in HiBiT at the endogenous locus of proteins with varying subcellular localization. Fixed CRISPR-modified clones or pools of cells were imaged by immunofluorescent staining using the Anti-HiBiT Monoclonal Antibody (red) and Hoechst dye (blue). Panel A. VCL-HiBiT pool. Panel B. SMARCA4-HiBiT clone. Panel C. HDAC2-HiBiT clone. Panel D. HSP90B1-HiBiT pool.
Affinity Tags
The most commonly used protein tags fall under the category of affinity tags. This means that the tag binds to another molecule or metal ion, making it easy to purify or pull down your protein of interest. In all cases, the tag will be fused to your protein of interest at either the amino (N) or carboxy (C) terminus by cloning into an expression vector. This protein fusion can then be expressed in cells or cell-free systems, depending on the promoter the vector contains.
It’s time to analyze your protein and you are trying to decide where to begin. You are asking questions like: Which protease do I choose? How much enzyme should I use in my digest? How long should I perform my digest?
Unfortunately, there is no one-size fits all answer to this type of question other than… “well it depends.” All protease digests will be a balance between denaturing the protein sample to allow access to cleavage sites, optimizing conditions for the protease to function, and compatibility with your workflow and downstream applications. We provide general guidelines that work for most samples, but frequently you will need to optimize the conditions need for your specific sample and application.
Here, I use the example of a trypsin digest for downstream mass spectrometry to highlight key questions to ask and factors that can be optimized for any digest.
Asp-N is a endoproteinase hydrolyzes peptide bonds on the N-terminal side of aspartic residues. The native form is isolated from Pseudomonas fragi. The majority of vendors currently provide a commercial product that consists of 2µg of lyophilized material in a flat bottom vial, and sold for $175–200 US. Formatting such a small amount of material in flat bottom vial can lead to inconsistent resuspension of the protease. Inconsistent working concentrations will lead to non-reproducible data. The current high price also prohibits large-scale use.
Are you looking for proteases to use in your research? Explore our portfolio of proteases today.
The new recombinant Asp-N protease is cloned from Stenotrophomonas maltophilia and expressed in E. coli. Recombinant Asp-N has similar amino acid cleavage specificity as compared to native Asp-N. Digestion of a yeast extract with native and recombinant Asp-N produces very similar results. Providing 10µg lyophilized material in V-shaped vial with a visible cake enables more consistent re-suspension resulting in reproducible data. Due to improved yields the list price is now approximately 40% less when compared to native enzyme. Learn more about this new recombinant Asp-N protease.
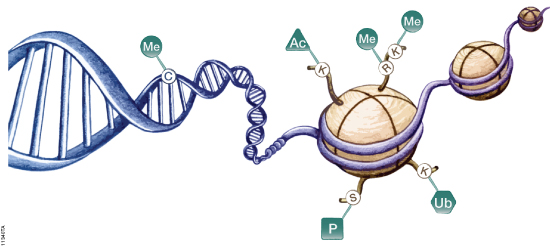
DNA is organized by protein:DNA complexes called nucleosomes in eukaryotes. Nucleosomes are composed of 147 base pairs of DNA wrapped around a histone octamer containing two copies of each core histone protein. Histone proteins play significant roles in many nuclear processes including transcription, DNA damage repair and heterochromatin formation. Histone proteins are extensively and dynamically post-translationally modified, and these post-translational modifications (PTMs) are thought to comprise a specific combinatorial PTM profile of a histone that dictates its specific function. Abnormal regulations of PTM may lead to developmental disorders and disease development such as cancer.
Recombinant erythropoietin (rhEPO) is often used as “doping agent” by athletes in endurance sports to increase blood oxygen capacity. Some strategies improve the pharmacological properties of erythropoietin (EPO) through the genetic and chemical modification of the native EPO protein. The EPO-Fcs are fusion proteins composed of monomeric or dimeric recombinant EPO and the dimeric Fc region of human IgG molecules. The Fc region includes the hinge region and the CH2 and CH3 domains. Recombinant human EPOs (rhEPO) fused to the IgG Fc domain demonstrate a prolonged half-life and enhanced erythropoietic activity in vivo compared with native or rhEPO.
Drug-testing agencies will need to obtain primary structure information and develop a reliable analytical method for the determination of EPO-Fc abuse in sport. The possibility of EPO-Fc detection using nanohigh-performance liquid chromatography−tandem mass spectrometry (HPLC−MS/MS) was already demonstrated (1). However, the prototyping peptides derived from EPO and IgG are not selective enough because both free proteins are naturally presented in human serum. In a recent publication, researchers describe the effort to identify peptides covering unknown fusion breakpoints (later referred to as “spacer” peptides; 2). The identification of “spacer” peptides will allow the confirmation of the presence of exogenous EPO-Fc in human biological fluids.
Are you looking for proteases to use in your research? Explore our portfolio of proteases today.
A bottom-up approach and the intact molecular weight measurement of deglycosylated protein and its IdeS proteolytic fractions was used to determine the amino acid sequence of EPO-Fc. Using multiple proteases, peptides covering unknown fusion breakpoints (spacer peptides) were identified.
Results indicated that “spacer peptides” could be used in the determination of EPO-Fc fusion proteins in biological samples using common LC−tandem MS methods.
Bottom-up proteomics focuses on the analysis of protein mixtures after enzymatic digestion of the proteins into peptides. The resulting complex mixture of peptides is analyzed by reverse-phase liquid chromatography (RP-LC) coupled to tandem mass spectrometry (MS/MS). Identification of peptides and subsequently proteins is completed by matching peptide fragment ion spectra to theoretical spectra generated from protein databases.
Trypsin has become the gold standard for protein digestion to peptides for shotgun proteomics. Trypsin is a serine protease. It cleaves proteins into peptides with an average size of 700-1500 daltons, which is in the ideal range for MS (1). It is highly specific, cutting at the carboxyl side of arginine and lysine residues. The C-terminal arginine and lysine peptides are charged, making them detectable by MS. Trypsin is highly active and tolerant of many additives.
Even with these technical features, the use of trypsin in bottom-up proteomics may impose certain limits in the ability to grasp the full proteome, Tightly-folded proteins can resist trypsin digestion. Post-translational modifications (PTMs) present a different challenge for trypsin because glycans often limit trypsin access to cleavage sites, and acetylation makes lysine and arginine residues resistant to trypsin digestion.
Are you looking for proteases to use in your research? Explore our portfolio of proteases today.
To overcome these problems, the proteomics community has begun to explore alternative proteases to complement trypsin. However, protocols, as well as expected results generated when using these alternative proteases have not been systematically documented.
In a recent reference (2), optimized protocols for six alternative proteases that have already shown promise in their applicability in proteomics, namely chymotrypsin, Lys-C, Lys-N, Asp-N, Glu-C and Arg-C have been created.
Data describe the appropriate MS data analysis methods and the anticipated results in the case of the analysis of a single protein (BSA) and a more complex cellular lysate (Escherichia coli). The digestion protocol presented here is convenient and robust and can be completed in approximately in 2 days.
Try a sample of high-efficiency Trypsin Platinum today!
Visit our website for more on Trypsin Platinum, Mass Spectrometry Grade, with enhanced proteolytic efficiency and superior autoproteolytic resistance.
Brachylophosaurus was a mid-sized member of the hadrosaurid family of dinosaurs living about 78 million years ago, and is known from several skeletons and bonebed material from the Judith River Formation of Montana and the Oldman Formation of Alberta. Recent fossil evidence indicates structures similar to blood vessels in location and morphology, have been recovered after demineralization of multiple dinosaur cortical bone fragments from multiple specimens, some of which are as old as 80 Ma. These structures were hypothesized to be either endogenous to the bone (i.e., of vascular origin) or the result of biofilm colonizing the empty network after degradation of original organic components (i.e., bacterial, slime mold or fungal in origin). Cleland et al. (1) tested the hypothesis that these structures are endogenous and thus retain proteins in common with extant archosaur blood vessels that can be detected with high-resolution mass spectrometry and confirmed by immunofluorescence.
Here we provide two examples of “atypical” experiments that take advantage of the properties of the ProteaseMAX™ Surfactant to improve studies involving digestion of complex protein mixtures.
Example 1 Clostridium difficile spores are considered the morphotype of infection, transmission and persistence of C. difficile infections. A recent publication (1) illustrated a novel strategy using three different approaches to identify proteins of the exosporium layer of C. difficile spores and complements previous proteomic studies on the entire C. difficile spores.
XWe use cookies and similar technologies to make our website work, run analytics, improve our website, and show you personalized content and advertising. Some of these cookies are essential for our website to work. For others, we won’t set them unless you accept them. To learn more about our approach to Privacy we invite you to Read More
By clicking “Accept All”, you consent to the use of ALL the cookies. However you may visit Cookie Settings to provide a controlled consent.
We use cookies and similar technologies to make our website work, run analytics, improve our website, and show you personalized content and advertising. Some of these cookies are essential for our website to work. For others, we won’t set them unless you accept them. To find out more about cookies and how to manage cookies, read our Cookie Policy.
If you are located in the EEA, the United Kingdom, or Switzerland, you can change your settings at any time by clicking Manage Cookie Consent in the footer of our website.
Necessary cookies are absolutely essential for the website to function properly. These cookies ensure basic functionalities and security features of the website, anonymously.
Cookie
Duration
Description
cookielawinfo-checbox-analytics
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Analytics".
cookielawinfo-checbox-functional
11 months
The cookie is set by GDPR cookie consent to record the user consent for the cookies in the category "Functional".
cookielawinfo-checbox-others
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Other.
cookielawinfo-checkbox-advertisement
1 year
The cookie is set by GDPR cookie consent to record the user consent for the cookies in the category "Advertisement".
cookielawinfo-checkbox-necessary
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookies is used to store the user consent for the cookies in the category "Necessary".
cookielawinfo-checkbox-performance
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Performance".
gdpr_status
6 months 2 days
This cookie is set by the provider Media.net. This cookie is used to check the status whether the user has accepted the cookie consent box. It also helps in not showing the cookie consent box upon re-entry to the website.
lang
This cookie is used to store the language preferences of a user to serve up content in that stored language the next time user visit the website.
viewed_cookie_policy
11 months
The cookie is set by the GDPR Cookie Consent plugin and is used to store whether or not user has consented to the use of cookies. It does not store any personal data.
Analytical cookies are used to understand how visitors interact with the website. These cookies help provide information on metrics the number of visitors, bounce rate, traffic source, etc.
Cookie
Duration
Description
SC_ANALYTICS_GLOBAL_COOKIE
10 years
This cookie is associated with Sitecore content and personalization. This cookie is used to identify the repeat visit from a single user. Sitecore will send a persistent session cookie to the web client.
vuid
2 years
This domain of this cookie is owned by Vimeo. This cookie is used by vimeo to collect tracking information. It sets a unique ID to embed videos to the website.
WMF-Last-Access
1 month 18 hours 24 minutes
This cookie is used to calculate unique devices accessing the website.
_ga
2 years
This cookie is installed by Google Analytics. The cookie is used to calculate visitor, session, campaign data and keep track of site usage for the site's analytics report. The cookies store information anonymously and assign a randomly generated number to identify unique visitors.
_gid
1 day
This cookie is installed by Google Analytics. The cookie is used to store information of how visitors use a website and helps in creating an analytics report of how the website is doing. The data collected including the number visitors, the source where they have come from, and the pages visted in an anonymous form.
Advertisement cookies are used to provide visitors with relevant ads and marketing campaigns. These cookies track visitors across websites and collect information to provide customized ads.
Cookie
Duration
Description
IDE
1 year 24 days
Used by Google DoubleClick and stores information about how the user uses the website and any other advertisement before visiting the website. This is used to present users with ads that are relevant to them according to the user profile.
test_cookie
15 minutes
This cookie is set by doubleclick.net. The purpose of the cookie is to determine if the user's browser supports cookies.
VISITOR_INFO1_LIVE
5 months 27 days
This cookie is set by Youtube. Used to track the information of the embedded YouTube videos on a website.
Performance cookies are used to understand and analyze the key performance indexes of the website which helps in delivering a better user experience for the visitors.
Cookie
Duration
Description
YSC
session
This cookies is set by Youtube and is used to track the views of embedded videos.
_gat_UA-62336821-1
1 minute
This is a pattern type cookie set by Google Analytics, where the pattern element on the name contains the unique identity number of the account or website it relates to. It appears to be a variation of the _gat cookie which is used to limit the amount of data recorded by Google on high traffic volume websites.