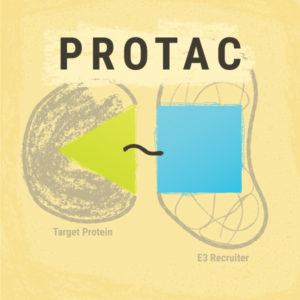
PROTACs or Proteolysis-Targeting Chimeras are an emerging tool in protein degradation studies, potentially suited to any need involving the removal of a specific protein. These small-molecule chimeras are exciting due to: 1) their target specificity; and 2) their ability to enable target destruction versus target inhibition. Here we discuss a paper that presents a roadmap for PROTAC development.
Destruction/Inhibition: Is There a Difference?
An analogy that microbiologists (and wrestlers or anyone that has ever spent time in a locker room shower) would understand, is fungicidal versus fungistatic compounds. A fungicidal compound kills fungus. A fungistatic compound just slows the fungus down a bit.
A small-molecule inhibitor attaches to its target protein, but for how long? What inhibitor testing must be done to determine how long the inhibition lasts?
On the other hand, a small-molecule agent that causes protein degradation first targets the protein of interest, then attaches ubiquitin to that target. Once a protein is marked with ubiquitin, it’s a dead man. E3 ligase must be involved, but if the ubiquitin is added by E3, the end is near. Next stop, Hades.
This ubiquitinated protein is headed to the proteasome and proteins that go there don’t come back. Ubiquitination was called the ‘molecular kiss of death’ when this discovery was awarded the Nobel prize in Chemistry in 2004.
About PROTACs
PROTACs are degrader molecules composed of three parts: 1) a ligand that is specific for the target protein; 2) a ligand for E3 ligase; and 3) a linker molecule that connects the two ligands. The E3 ligase is one of three enzymes that can add ubiquitin to a cellular component, but only ubiquitins added by the E3 ligase cause targeting to the proteasome (Zoppi et al.).
A Recent PROTAC Report
Don’t PROTACs sound fantastic in their protein destruction abilities? Let’s make one for every protein we want to destroy, and cure cancer next month, right?
If only it was that simple. While the technology holds great promise as a therapeutic method, as Zoppi et al. note in their late 2018 Journal of Medicinal Chemistry paper:
“To fulfill the potential of targeted protein degradation, a general methodology for an efficient PROTAC design would be desirable. However, the development of active PROTAC degraders is often a laborious and unguided process.”
In developing PROTAC degrader molecules there are choices to make for each of the three parts, and thus multiple variables to optimize. Considerations for the linker alone include length, chemical composition and site or attachment (1). Additionally, there are a number of human E3 ligases, but only a few of them have good quality ligands that have been used successfully in PROTACs. PROTACs made with a given protein target ligand and different E3 ligase ligands exhibit differences in degradation efficacy.
Roadmap for PROTAC Development: An Iterative Design Approach
In their J. Med. Chem. paper, “Iterative Design and Optimization of Initially Inactive Proteolysis Targeting Chimeras (PROTACs) Identify VZ185 as a Potent, Fast, and Selective von Hippel–Lindau (VHL) Based Dual Degrader Probe of BRD9 and BRD7”, Zoppi et al. developed probe-quality PROTACs from a ligase-target pair that had previously seemed incompatible, using VHL and the protein BRD9. BRD9 and its homolog BRD7 are bromodomain-containing subunits of chromatin-remodeling complexes.
BRD9 and BRD7 have been popular targets of cancer immunotherapy and potent inhibitors of these bromodomains have emerged from structure-guided medicinal chemistry campaigns.
The authors carefully examined crystal structures of compounds to identify suitable attachment and ligand-binding sites, examined binding biophysically and structurally, and evolved their high-quality (probe-quality) degrader over three generations. They examined and tested PROTACs in several cell types, with numerous E3 ligands, using various linker lengths and by examining structure and binding percentages by protein, as shown in Tables 2 and 3 of this open-access paper.
The meticulous iterative optimization performed in this paper resulted in a probe-quality degrader VZ185. The authors describe VZ185 as
“a potent, fast and selective degrader of BRD9 and its close homolog BRD7”.
The authors note that these findings provide not only a new chemical tool for BRD7/9 degradation but also
“a roadmap for PROTAC development against seemingly incompatible target-ligase combinations”.
Promega Scientists and Products Involved in This Research
We are particularly proud of this work because two of the J. Med. Chem. authors, Danette Daniels and Kristin Riching, are R & D scientists here at Promega. Other collaborators include the Dundee laboratory.
The following reagents used in this research are available from Promega:
CellTiter-Glo Assays were used to determine cell proliferation.
Nano-Glo HiBiT Lytic Detection System was used to determine endogenous tagging and kinetic degradation.
The extended time-release substrate Nano-Glo Endurazine was added to cells before exposure to their degrader VZ185 compound.
Multiwell plates were read on a GloMax® Discover.
Here is the paper:
Zoppi, V. et al. (2018) Iterative Design and Optimization of Initially Inactive Proteolysis Targeting Chimeras (PROTACs) Identify VZ185 as a Potent, Fast, and Selective von Hippel–Lindau (VHL) Based Dual Degrader Probe of BRD9 and BRD7 J. Med Chem.
2018 Dec 12. doi: 10.1021/acs.jmedchem.8b01413. [Epub ahead of print]
Learn more about targeted protein degradation and PROTACS here.
Related Posts
Kari Kenefick
Latest posts by Kari Kenefick (see all)
- What Drives Muscle Fiber Shifts in Obesity and Type ll Diabetes? - October 9, 2025
- How Thalidomide and Molecular Glues Are Redefining Drug Discovery - August 21, 2025
- ATP-Powered Proteins Beyond Kinases – and Why Helicases Are Stealing the Spotlight - August 7, 2025
One thoughtful comment