
While PROTACs might not be the topic of conversation at high society cocktail parties, or merit cover stories in glamor magazines, they’re certainly shaking up the drug discovery industry. PROTAC® degraders, together with related compounds like molecular glues and LYTACs, are the basic tools for a targeted protein degradation strategy. Research in this field is advancing rapidly, enabling the development of therapies for disease targets disease targets previously thought to be “undruggable”. This blog post provides an overview of PROTACs based on frequently asked questions.
What is a PROTAC?
The acronym PROTAC stands for proteolysis-targeting chimera. As the name implies, these molecules contain multiple functional units and are used to target specific proteins within the cell for degradation by the cell’s garbage disposal system—the ubiquitin-proteasome system or UPS. A typical PROTAC consists of two functional, protein-binding units or ligands separated by a linker.
How do PROTACs work?
The bifunctional nature of a PROTAC is critical to the way it works within the cell. One of the PROTAC ligands (sometimes called the “warhead”) binds to the target protein of interest. The other ligand (the “anchor”) binds to a component of the UPS machinery—typically, a protein that recruits an E3 ubiquitin ligase. Simultaneous binding of these two proteins by the PROTAC results in the formation of a ternary complex. Once this complex is formed, an E3 ligase catalyzes the sequential addition of ubiquitin residues to the target protein. This ubiquitin chain attracts the attention of the UPS, and the target protein is subsequently degraded.
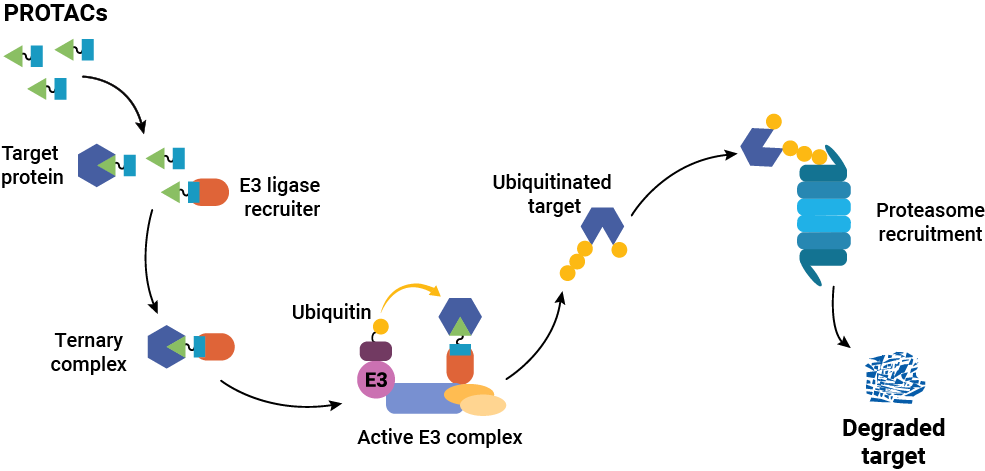
It’s important to note that the PROTAC is released after ubiquitination of the target protein, so it is essentially recycled in the process. Therefore, a relatively small amount of PROTAC can catalyze the degradation of a large amount of target protein.
Why are PROTACs important?
Once a target protein is identified as being associated with a disease, traditional drug discovery strategies have centered around finding an inhibitor to that protein. In many cases, such an approach involves screening a library of small-molecule drug candidates to identify those that bind to an active site on the target protein and modulate its activity. This approach of “occupancy-driven pharmacology” has its limitations (1), especially for proteins that do not have enzymatic activity. As a result, an estimated 85% of known protein targets remain undruggable (2). Further, even in cases where suitable inhibitors exist, high concentrations of these drugs are often required to achieve therapeutic effects (3).
PROTACs overcome the disadvantages of inhibitor-driven approaches to drug development. They can be designed to bind multiple sites on most protein targets, and their catalytic mechanism of action allows for reversible, concentration-dependent modulation of the target protein level. In some situations, especially when studying complex signaling pathways, degradation of a target protein may offer an advantage over merely inhibiting its activity.
How were PROTACs discovered?
The first PROTAC reported was a peptide synthesized and tested by the laboratories of Craig Crews and Raymond Deshaies in 2001 (4). In this proof-of-concept study, the PROTAC was designed to target the enzyme methionine aminopeptidase-2 (MetAP-2) to a component of the ubiquitin ligase complex. The study showed that Met-AP2 was ubiquitinated and degraded in a PROTAC-dependent manner, setting the stage for future PROTAC development. Further applications of peptide-based PROTACs followed, but these molecules were often hampered by poor cell permeability (reviewed in 5).
Successful application of the first small-molecule PROTAC was published in 2008 (6). This molecule displayed good cell permeability and successfully targeted the androgen receptor for degradation in a PROTAC concentration-dependent manner, suggesting applications in prostate cancer characterized by increased androgen receptor expression.
Since those early discoveries, PROTAC research has experienced exponential growth (reviewed in 7). At present, PROTAC development offers an exciting alternative to the traditional inhibitor-based approach to small-molecule drug discovery. According to PROTAC-DB, a web-accessible database that tracks PROTAC development, there were 2,258 PROTACs in development as of April 2021.
How do I design an effective PROTAC?
Multiple factors must be considered in the rational design of a PROTAC. While structural studies can provide insight into the binding site for targets of interest from crystallographic data, these data are not available for many targets or potential PROTACs. Accordingly, rational PROTAC design is driven by examining the mechanistic aspects of each stage of the targeted protein degradation process. General considerations for PROTAC design have been reviewed by Paiva and Crews (8). Leissing et al. presented an overview of structure-guided design principles, with an emphasis on ternary complex formation (9). Another review discussed structure-guided PROTAC design to target chromatin remodeling complex components implicated in several types of cancer (10).
Typically, PROTAC design focuses on each component of the molecule: the target-binding ligand or “warhead”, the linker, and the E3 ligase-recruiting ligand or “anchor”.
Target-Binding Ligand
In most cases, the PROTAC warhead will have a high affinity for its target protein. Design strategies can take advantage of known small-molecule inhibitors that have been extensively characterized and are specific to the target of interest. However, ligands that bind multiple targets may offer advantages, and even those with weak affinity for the target protein may be stabilized upon ternary complex formation, leading to efficient target degradation (11).
An interesting finding in PROTAC development is the use of trivalent PROTACs (12). These molecules include two ligands that bind the target protein, and this increased avidity for the target results in greater stabilization of the ternary complex and increased target degradation.
Linker
A PROTAC linker is often overlooked during the design stage, and its importance may be underestimated. Increasingly, more complex and functional linkers are being used, with advantages in ternary complex stabilization and overall improvements in degradation efficiency (reviewed in 13). Both linker composition and linker length play important roles in optimizing PROTAC efficiency (14).
E3 Ligand
Over 600 E3 ubiquitin ligases are known; however, most E3 ligase-recruiting ligand development is targeted at just a handful of these enzymes (15). Of these, the most common are the von Hippel-Lindau (VHL) E3 ligase and cereblon (CRBN). An exhaustive review provides insight into the synthetic pathways available when designing ligands that bind to these and other E3 ligases (15). The review also includes a tool to assist with selecting a suitable ligand, as well as a list of popular, commercially available building blocks for developing E3 ligands.
How can I test if a PROTAC will work in live cells?
A variety of biochemical assays exist to examine each step in the targeted protein degradation process; however, traditional methods typically provide end-point results and are not suitable for high-throughput analysis. Live-cell assays to monitor degradation in real time offer significant advantages (16). Of these assays, NanoBRET™-based methods offer sensitive and convenient measurement of PROTAC kinetics, as well as a broader understanding of the complex degradation processes in live cells (17). NanoBRET™ VHL and CRBN Ternary Complex Starter Kits provide a convenient method to get started with assays to measure ternary complex formation and simultaneously quantitate target protein degradation.
Are PROTACs being used in clinical applications?
The first PROTAC to enter clinical trials, in 2019, was developed by Arvinas, Inc. for the treatment of prostate cancer. In February 2022, the company reported interim results stating that the PROTAC, ARV-110 or bavdegalutamide, “continues to provide evidence of anti-tumor activity and patient benefit in metastatic castration-resistant prostate cancer (mCRPC)”. In collaboration with Pfizer, Arvinas has also reported encouraging initial results for another PROTAC, ARV-471, in the treatment of breast cancer. Other types of cancer are being targeted by PROTACs and related degraders (reviewed in 18, 19). A promising development in preclinical studies is the use of PROTACs to target KRAS-driven cancers, which have historically been difficult to target effectively (20).
Are you interested in PROTAC drug discovery but don’t know where to start? Our Compound Profiling Services can help.
Want to learn more? We have many posts about targeted protein degradation right here at Promega Connections. Check them out.
PROTAC is a registered trademark of Arvinas Operations, Inc.
References
- Valeur, E. and Jimonet, P. (2018) New modalities, technologies, and partnerships in probe and lead generation: enabling a mode-of-action centric paradigm. J. Med. Chem. 61, 9004–9029.
- Pathmanathan, S. et al. (2022) Drugging the undruggable proteins in cancer: A systems biology approach. Curr. Opin. Chem. Biol. 66, 102079.
- Adjei, A.A. (2006) What is the right dose? The elusive optimal biologic dose in phase I clinical trials. J. Clin. Oncol. 24, 4054–4055.
- Sakamoto, K.M. et al. (2001) Protacs: Chimeric molecules that target proteins to the Skp1–Cullin–F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 98(15), 8554–8559.
- Pettersson, M. and Crews, C. (2019) PROteolysis TArgeting Chimeras (PROTACs)—Past, present and future. Drug Discov. Today Technol. 31, 15–27.
- Schneekloth, A.R. et al. (2008) Targeted intracellular protein degradation induced by a small molecule: En route to chemical proteomics. Bioorganic Med. Chem. Lett. 18, 5904–5908.
- Békés, M. et al. (2022) PROTAC targeted protein degraders: the past is prologue. Nat. Rev. Drug Discov. 21,181–200.
- Paiva, S.-L. and Crews, C. (2019) Targeted protein degradation: elements of PROTAC design. Curr. Opin. Chem. Biol. 50, 111–119.
- Leissing, T.M. et al. (2020) Structure driven compound optimization in targeted protein degradation. Drug Discov. Today Technol. 37, 73–82.
- Farnaby, W. et al. (2019) BAF complex vulnerabilities in cancer demonstrated via structure-based PROTAC design. Nat. Chem. Biol. 15, 672–680.
- Bondeson, D.P. et al. (2018) Lessons in PROTAC design from selective degradation with a promiscuous warhead. Cell Chem. Biol. 25, 78–87.
- Imaide, S. et al. (2021) Trivalent PROTACs enhance protein degradation via combined avidity and cooperativity. Nat. Chem. Biol. 17, 1157–1167.
- Troup, R.I. et al. (2020) Current strategies for the design of PROTAC linkers: a critical review. Explor. Target Antitumor Ther. 1, 273–312.
- Zagidullin, A. et al. (2020) Novel approaches for the rational design of PROTAC linkers. Explor. Target Antitumor Ther. 1, 381–390.
- Bricelj, A. et al. (2021) E3 ligase ligands in successful PROTACs: an overview of syntheses and linker attachment points. Front. Chem. 9, 707317.
- Daniels, D.L. et al. (2019) Monitoring and deciphering protein degradation pathways inside cells. Drug Discov. Today Technol. 31, 61–68.
- Riching, K.M. et al. (2018) Quantitative live-cell kinetic degradation and mechanistic profiling of PROTAC mode of action. ACS Chem. Biol. 13, 2758−2770.
- Mullard, A. (2021) Targeted protein degraders crowd into the clinic. Nat. Rev. Drug Disc. 20, 247–250.
- Qi, S.-M. et al. (2021) PROTAC: an effective targeted protein degradation strategy for cancer therapy. Front. Pharmacol. 12, 692574.
- Zhou, C. et al. (2022) Discovery of the first-in-class agonist-based SOS1 PROTACs effective in human cancer cells harboring various KRAS mutations. J. Med. Chem. 65, 3923−3942.
Latest posts by Ken Doyle (see all)
- CRISPR/Cas9 Knock-In Tagging: Simplifying the Study of Endogenous Biology - November 18, 2025
- Will Artificial Intelligence (AI) Transform the Future of Life Science Research? - February 1, 2024
- RAF Inhibitors: Quantifying Drug-Target Occupancy at Active RAS-RAF Complexes in Live Cells - September 5, 2023