Analyzing more than one cellular biomarker (multiplexing) in a single sample is advantageous for a number of reasons. Multiplexing allows researchers to save money and time, while conserving critical samples. In addition, understanding the relationship between cell biomarkers can provide a more complete picture of cell health, leading to improved predictive models for drug discovery. Understanding biomarker relationships can also minimize ambiguity in the data set and validate if a treatment effect is real or an artifact of the system. To avoid repeat experiments and extract the most physiologically relevant data from multiplex cell-based assays, we discuss considerations around assay choices, cell type, cell culture, treatment parameters, detection and appropriate experimental controls.
Assay Choices
Select cell-based assays with relevance to treatment outcomes you are studying such as cell viability, apoptosis and cytotoxicity. (See this blog for a list of Promega cell-based assays and the outcomes that they measure). A multiplex cell-based assay should meet the following criteria:
- Assay volumes must fit in the volume of the assay well or be easily physically separated.
- Signal readouts from each assay must be spectrally distinct.
- Assay chemistries must be compatible (e.g., avoid components that might precipitate or interfere with assay signal)
- Assays using nonlytic reagents should be performed before introducing assays with cell lysis reagents. This may seem obvious, but reviewing the principle behind the assay chemistries will help map out your multiplex procedure.
- You may need to use a more concentrated assay working reagent for the first assay to leave enough room in the well to add the appropriate volume of the second assay reagent. Consult the assay manufacturer’s technical information for any instructions to concentrate their assay reagent.
- When combining signal readouts, it is not practical to perform colorimetric assays in the same well as fluorescent or luminescent assays due to the ability of the chromogen to quench these signals. If the sample can be physically divided or separated (culture medium vs cells) into different wells, then the assay chemistries will not interfere with each other.
- Combining multiple fluorescent assays should be possible so long as 1) the readouts are spectrally distinct, 2) there is no resonance transfer 3) there are no quenching agents (e.g., detergents, redox) affecting companion fluors, and 4) there is minimal autofluorescence.
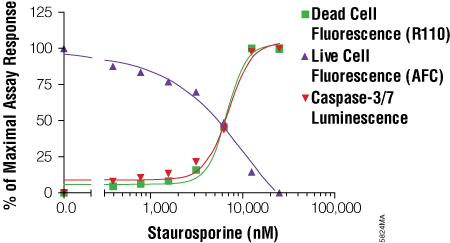
- Promega has had great success in combining cell-based assays with fluorescent and luminescent readouts. The multiplex assay format typically begins with fluorescent assays (nonlytic) followed by luminogenic assays (lytic). An example of this format is the ApoTox-Glo™ Triplex Assay where the detection of multiple biomarkers occurs sequentially.
- Combining two or more luminescent-based assays can be difficult. Promega provides options for multiplexing luminescent assays.
- The Nano-Glo® Dual-Luciferase assay takes best advantage of the power of NanoLuc® luciferase and firefly luciferase, providing an assay that enables assay design where either NanoLuc or firefly luciferase can be the primary reporter, assay design with two primary reports to measure two different biological responses simultaneously, or coincidence reporter assays in which both reporters are expressed from a single open reading frame.
- The Dual-Luciferase® Reporter Assay System offers a multiplex format that uses two unique bioluminogenic enzymes, firefly (Photinus pyralis) and Renilla (Renilla reniformis or sea pansy) luciferases. The signals are read sequentially after quenching the initial firefly luciferase reaction. Therefore, look for luminescent assay chemistries that incorporate different luminogenic enzymes and include quenching agents to prevent signal overlap when multiplexing.
Cell Type
The cell type can greatly affect the experimental design, results and interpretation. Determine if the cell line is amenable to the multiplex assay format and consider the following:
- When using established cell lines such as Hep-G2 and PC-3 cancer cells, viability (live cell) is generally higher compared to primary cell cultures (e.g., HUVEC, hepatocytes).
- Conversely, cytotoxicity markers (dead cell) are seen at higher levels in primary cultures due to the higher spontaneous rate of death compared to stable cell lines.
- Primary cells may provide a more biologically relevant model for compound testing, but they may require a significant amount of time to optimize assay conditions given the lifespan of these cells.
Cell Culture

The complex nature of culturing cells and keeping them healthy can affect the reproducibility and reliability of the data generated in multiplex assays. It is no secret that cell-based assays have greater variability than biochemical-based assays because biochemical assays have fewer components (e.g., test compound, enzyme and activators). Minimizing variables introduced into the cell culture media and how the cells are handled will help improve the quality of the multiplex data.
- The biggest culprit in variable results is the supplemental serum, usually bovine, added to the media. Always qualify a new lot of serum from the same vendor (e.g., assess cell-growth behavior with new serum lot). Use a single serum lot if possible when performing large or lengthy studies. The alternative is to switch to a serum-free medium to avoid the effect serum has on cell health and subsequently, assay results.
- Use low-passage numbers. Frozen cell stocks should be grown to an optimal low-passage number (e.g., split cells 20–22 times) that ideally offers cells at their peak metabolic condition. Cells of a higher passage number can change characteristics such as expression or suppression of a given phenotype or show decreased viability. Produce a sufficient cell amount at the low-passage number then freeze aliquots for each experiment, so that passage number is consistent.
- Verify the cell line identity. If cell growth or cell response to treatment differs over time, this may indicate the need to verify cell line identity as well. For more information visit our cell line authentication guide.
- Trypsinization of adherent cells should be optimized since excess trypsin can kill cells and too little trypsin causes cell clumping. Inconsistent detachment causes incorrect cell number calculations that will affect assay results.
- Use trypan blue exclusion to assure viable cells are plated into assay wells.
- Transfection efficiency should be assayed to assure consistent expression of the reporter or biomarker between cell cultures.
- Avoid pipetting inaccuracies. Highly variable results are seen when cells are not kept evenly suspended in the pipette during well transfer or aliquot freezing. Gently pipette a cell culture up and down the same number of times from well-to-well before transferring. Rough pipette handling of cells can kill them, causing poor viability and false cytotoxicity.
- Any biological contamination (e.g., mycoplasm, mold, bacteria, other cell lines) can alter assay response. Poor reproducibility of your results may signal contamination. Discard contaminated cultures if there is any doubt to eliminate ambiguity in your results.
- Optimize cell density. Sparse cell cultures dispensed into wells can show higher detection sensitivity compared to high-density plated cells. Higher cell density or overgrown cultures can show higher background signals and also may require longer exposure times to the test compound for any measureable effect. To optimize cell density, generate a standard curve (serial dilutions) of plated cell numbers and assay for viability and cytotoxicity. This will help you determine when cells should be treated and the multiplex assay performed. This will help determine the sensitivity and range of the assay system as well before test compounds are introduced.
Treatment Parameters
All assay parameters can have an impact on multiplex results but dosage of the test compound and timing of the multiplex assay for a given cell line have an enormous effect on multiplex assay success.
- Timing is everything. Pay close attention to when you need to perform the multiplex cell-based assay for a given cell line. The half-life of the biomarkers can affect the accuracy of your assay results (e.g., are you able to detect the biomarker or not at a given time point). Testing multiple time points when generating dose response curves (time-course study) may be cumbersome but well worth the quality of information for dosing and treatment scheduling.
- Understanding the relationship between two or more biomarkers and when they appear (prior to treatment) helps make multiplex assays more robust. For example, if a decrease in the viability marker does not show a concomitant increase in the cytotoxicity marker, then we may theorize that 1) one of the assays is affected by the test compound in some way, 2) cytostasis has occurred (live cells not dividing) or 3) the time point for detecting the cytotoxicity biomarker has passed and the assay should be repeated using a shorter exposure time point. Interpretation of the data can be strengthened by performing an multiplex cell-based assay.
Detection and Measurement
A multiplex cell-based assay can be affected by the detection instruments (fluorometers, luminometers and multimode plate readers) and assay consumables used. Here are some variables to consider when you wish to improve sensitivity or dynamic range of your multiplex assay.
- Filter sets. Not observing a change in the fluorescent signal as you increase the cell number for treated and nontreated replicates may indicate an incorrect filter set. Compare the peak excitation and emission spectra of each fluorophore against the instrument filter set range. Make sure there is at least a 50% overlap in the wavelength range of the filter with the fluor excitation and emission maxims.
- Instrument gain. If you see very large error bars between treated and nontreated replicates, you may have the gain set too low.
- Microplates. White, opaque plates provide higher signals for fluorescent or luminescent readouts and may reduce the upper limit of the multiplex assay exhibiting saturation. However, sensitivity may not increase since there will be higher backgrounds as well. Black, opaque plates have lower signal and background responses, but very little cross-talk or light scattering compared to white, opaque plates. If a luminescent signal varies more than 2 logs between replicates, then use black, opaque plates to limit crosstalk. Clear bottom plates allow you to view cell morphology prior to the assay but can have more crosstalk. We have more information on choosing the best multi-well plate for your multiplex cell-based assay.
- Well-to-well crosstalk and other instrument artifacts. Be sure to use an instrument that minimizes problems like cross talk and provides you with the dynamic range and sensitivity you need.
Assay Controls
Assay Controls: Controls are necessary to let you know if the multiplex assay is functioning properly and to normalize data against variables across all the samples.
- No-Cell Control defines the background of the assay (e.g., media, plate, reagent, detector) and is subtracted as background in data processing.
- Untreated-Cells Control shows any vehicle effects and also is used in analysis to determine fold response (e.g., 100% viability, 0% cytotoxicity) for relative response ratio.
- Positive Control uses a known inducer of a response (e.g., cytotoxicity, apoptosis), giving insight into pathway effects. It is also used in to determine fold response (e.g., 100% viability, 0% cytotoxicity) for relative response ratio.
- Multiplate Assay Controls are replicates of Untreated-Cells Controls and Positive Controls. It demonstrates any effects from assay processing, temperature, timing, etc.
Multiplexing cell-based assays gives you greater confidence in the biomarkers you are measuring by putting them in a more biologically relevant context. Practicing good cell culture techniques and optimizing multiplex assay parameters will improve data quality and reliability in your research activities.
First published in June 2011. Updated June 18, 2024.