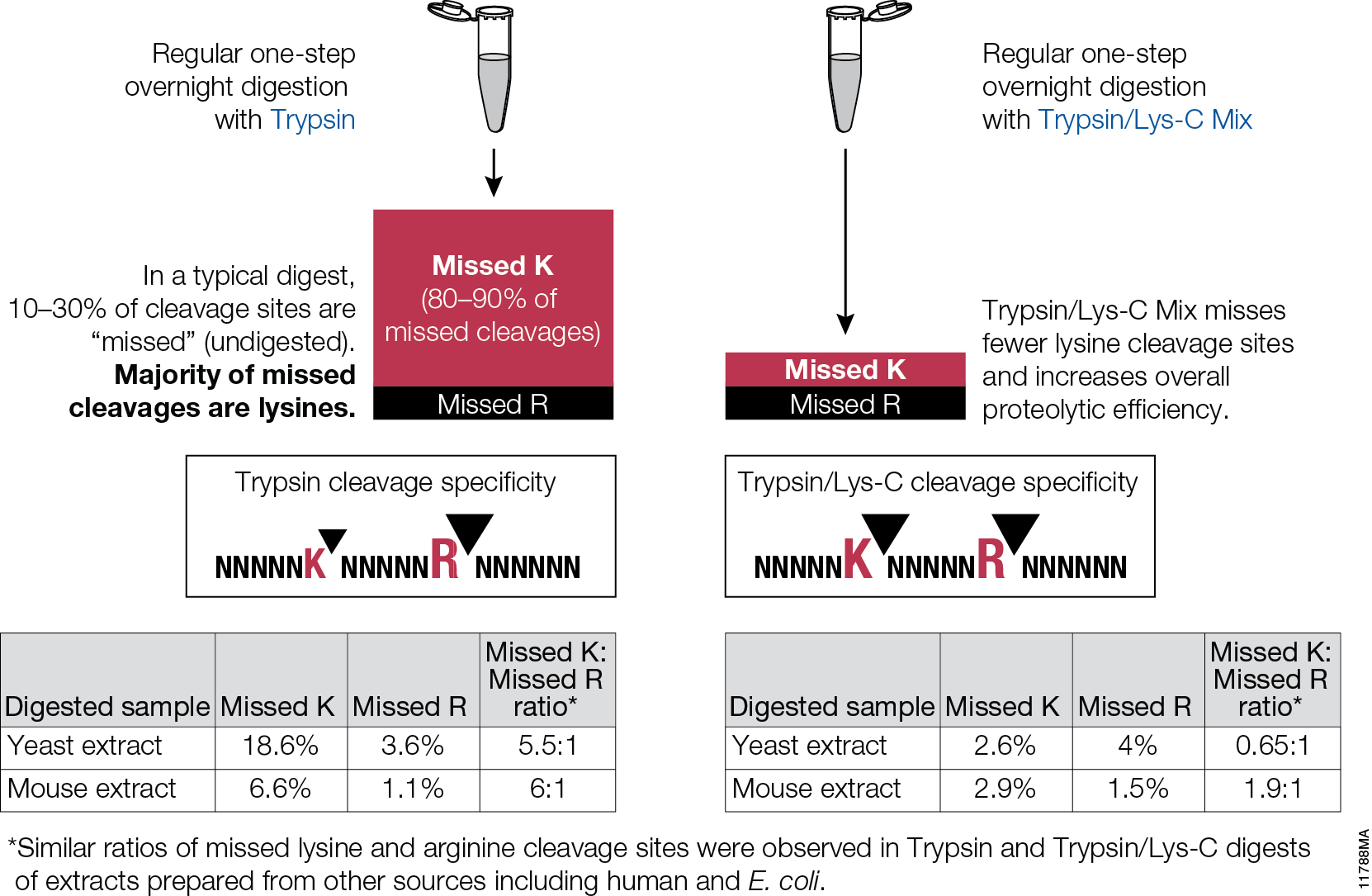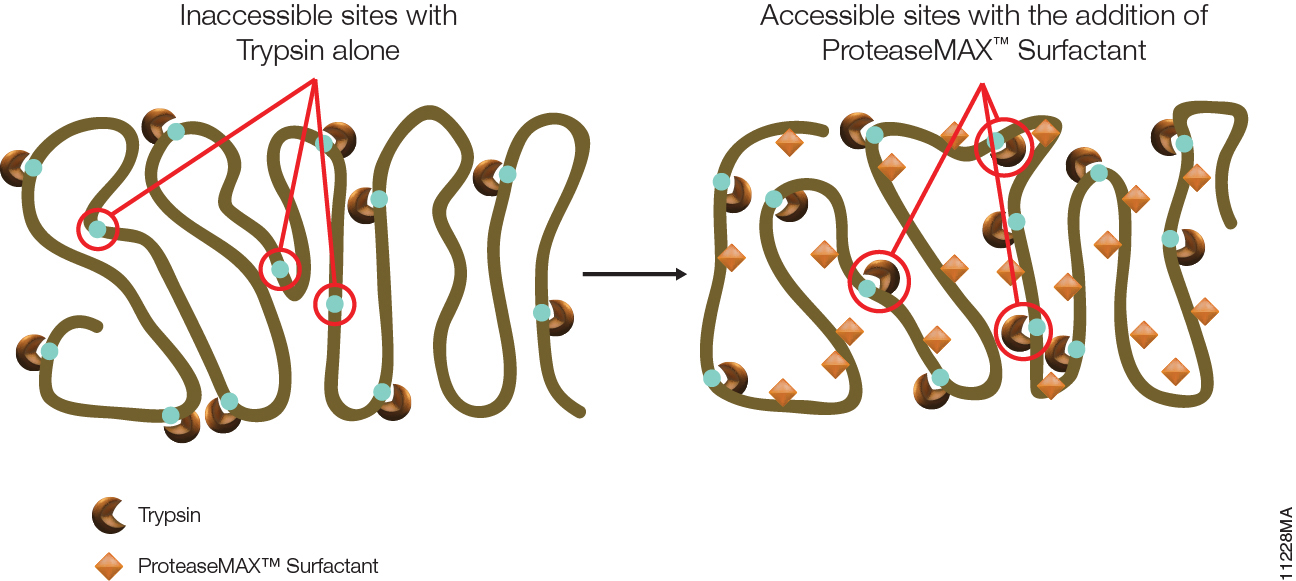
Post-translational modifications (PTM) of proteins are essential for the function of many proteins, but aberrant modification of protein residues also can interfere with protein function. PTMs occur in two ways. Proteins may be modified through the activity of enzymes such as kinases, phosphorylases, glycosylases and others that add or remove specific chemical moieties to amino acid residues. PTMs can also result from non-enzymatic reaction between electrophilic compounds and nucleophilic arginine and lysine residues within a protein. Metabolites and metabolic by products produced during glycolysis, especially glyoxal and methylglyoxal (MGO), are examples of such electrophilic compounds. These compounds can react with arginine and lysine to produce advanced glycation end products (AGEs), which are biomarkers associated with aging and degenerative diseases such as Alzheimer disease, diabetes and others. MGO is also elevated in tumors that have switched from oxidative phosphorylation to glycolysis as their main energy production pathway.
Only limited information is available about site-specific MGO PTMs in mammal cells, and most studies have focused on measuring the amount of MGO modifications in a treatment scenario compared to a control. Donnellan and colleagues recently published work to identify specific MGO protein modifications. They used a “bottom-up” proteomic analysis of WIL2-NS B lymphoblastoid whole-cell lysates to identify specific MGO-modified proteins. In particular, the group was looking for modifications in proteins that might explain how MGO activity contributes to aneuploidy.
For the study, 100µg of cellular protein extract was reduced with dithiothreitol and then alkylated with chloroacetamide. The sample was diluted to reduce urea concentration. Trypsin Gold was added and samples were digested for 8 hours at 37°C. Digestion was terminated by adding formic acid. For ProAlanase digestion, 20µg of protein was reduced, alkylated and diluted to reduce urea concentration before adding digesting with ProAlanase for 4 hours at 37°C.
Are you looking for proteases to use in your research?
Explore our portfolio of proteases today.
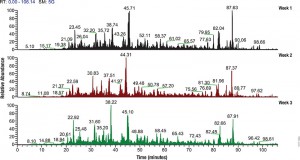
The authors identified 519 MGO-modified proteins. Most of the modifications were identified in the trypsin digestion reactions; however, ProAlanase digestion increased the number of MGO modifications identified by approximately 25% (with less than 4% of the modification sites being detected in both the ProAlanase and trypsin digestion reactions. The authors suggest that ProAlanase increased sequence coverage to reveal sites not detected in the trypsin digestions. Therefore, they conclude that ProAlanase can be used along with trypsin digestion to increase the identification of MGO modifications.
ProAlanase can be used along with trypsin digestion to increase the identification of MGO modifications.
MGO-modified proteins from the WIL2-NS whole cell lysates included proteins involved in glycolysis, translation initiation, protein folding, mRNA splicing, cell-to-cell adhesion, heat response, nucleosome assembly, protein SUMOylation and the G2/M cell cycle transition. More work to further characterize the sites of these modifications and their potential effects on the function of the modified proteins is ongoing.
Read more about ProAlanase, a new site-specific endoprotease from Promega.
Literature Cited
Donnelian, L et al. (2022) Proteomic Analysis of Methylglyoxal Modifications Reveals Susceptibility of Glycolytic Enzymes to Dicarbonyl Stress Int. J. Mol. Sci. 23(7), 3689 doi.org/10.3390/ijms23073689
 Filter-aided sample preparation (FASP) method is used for the on-filter digestion of proteins prior to mass-spectrometry-based analyses (1,2). FASP was designed for the removal of detergents, and chaotropes that were used for sample preparation. In addition, FASP removes components such as salts, nucleic acids and lipids. Akylation of reduced cysteine residues is also carried out on filter, after which protein is proteolyzed by use of trypsin on filter in the optimal buffer of the enzyme. Subsequent elution and desalting of the peptide-rich solution then provides a sample ready for LC–MS/MS analysis.
Filter-aided sample preparation (FASP) method is used for the on-filter digestion of proteins prior to mass-spectrometry-based analyses (1,2). FASP was designed for the removal of detergents, and chaotropes that were used for sample preparation. In addition, FASP removes components such as salts, nucleic acids and lipids. Akylation of reduced cysteine residues is also carried out on filter, after which protein is proteolyzed by use of trypsin on filter in the optimal buffer of the enzyme. Subsequent elution and desalting of the peptide-rich solution then provides a sample ready for LC–MS/MS analysis.