In late May 2026, a clinical trial result landed in the New England Journal of Medicine and immediately rewrote what oncologists believed was possible for patients with metastatic pancreatic cancer. Before the paper was published, people in the field were already calling it “transformative.” The data, when it came, agreed. In a disease where most second-line treatments offer months at best, a drug called daraxonrasib nearly doubled how long patients lived compared to those who received chemotherapy.¹
RAS proteins, which regulate cell growth and are mutated in more than a third of all human cancers,² had spent forty years resisting every attempt to drug them. The protein’s surface offered no obvious foothold for a small molecule. Once the word “undruggable” attached itself to the RAS protein family, most of the field moved on to more cooperative targets.
Some researchers stayed. And Promega stayed committed to the question that never goes away: does this new compound work inside a living cell? When the next chapter of the RAS story arrived, the tools were ready. Daraxonrasib is one culmination of a much longer story, one that matters for every researcher pursuing a target the field has written off.
The First Answer
To understand what daraxonrasib represents, it helps to see how an earlier chapter of the RAS story faced the same fundamental measurement challenge.
Understanding the expression, function and dynamics of
proteins in their native environment is a fundamental goal that’s common to
diverse aspects of molecular and cell biology. To study a protein, it must
first be labeled—either directly or indirectly—with a “tag” that allows
specific and sensitive detection.
Using a labeled antibody to the protein of interest is a
common method to study native proteins. However, antibody-based assays, such as
ELISAs and Western blots, are not suitable for use in live cells. These
techniques are also limited by throughput and sensitivity. Further, suitable
antibodies may not be available for the target protein of interest.
Targeted protein degradation (TPD) is an emerging drug discovery strategy that offers an entirely different approach to tackle disease-relevant proteins, including classic “undruggable” targets. Instead of inhibiting protein function, small molecules like PROTACs and molecular glues co-opt the cell’s own ubiquitin-proteasome system to eliminate specific proteins altogether. But as this targeted approach gains traction, it also challenges existing methods for validating compound activity.
How do you confirm that degradation is happening in a biologically-relevant system? Can you validate protein degradation in real-time?
The Wnt/β-catenin pathway, long studied in the context of developmental biology, has become increasingly recognized for its role in a wide range of human diseases. Its dysregulation has been implicated in cancer, fibrosis, immune modulation, and neurodegenerative conditions—making it a clinically actionable target across diverse therapeutic areas1. In this blog, we cover the fundamentals of Wnt/β-catenin signaling, highlight ongoing research efforts to understand its role in disease, and show how combining live-cell imaging with luminescent assays complements functional studies.
The human body has an incredible capacity for self-repair. Our skin can regenerate after a small cut, bones can heal after fractures and even the liver can regrow to its original size after 70% is lost or removed (3). However, when it comes to the heart, the story is very different. As Miley Cyrus once sang, “nothing breaks like a heart” – and science agrees. Unlike other organs, the heart has almost no ability to regenerate itself after injuries. In instances like myocardial infarctions, more commonly known as heart attacks, large amounts of cardiomyocytes (CMs)—the cells responsible for heart muscle contraction—are lost and cannot be regenerated, causing the formation of non-regenerative fibrotic scar tissue and, ultimately, decline in heart function (1).
Post-translational modifications of proteins are critical for proper protein function. Modifications such as phosphorylation/dephosphorylation can act as switches that activate or inactivate proteins in signaling cascades. The addition of specific sugars to membrane proteins on cells are critical for recognition, interaction with the extracellular matrix and other activities. While we know volumes about some types of protein modifications, ADP-ribosylation on aspartate and glutamate residues has been more difficult to study because of the chemical instability of these ester-linked modifications.
Matić Lab (Eduardo José Longarini and Ivan Matić) recently published a study that explored mono-ADP-ribosylation (ADPr) on aspartate and glutamate residues by the protein PARP1 and its potential reversal by PARG. PARP1 and PARG signaling are central to DNA repair and apoptosis pathways, making them potentially powerful therapeutic targets in cancer or neurodegenerative diseases in which DNA repair processes are often disrupted.
Fluorescent tags (fluorophores), have become excellent tools for labeling cells and cellular components. They can be used for imaging large molecules like proteins, on down to cellular components and enzymes such as transcription factors. Once labeled, these molecules can be tracked in tissue or inside a cell, when the right tag is used.
What is the ‘right’ tag? It’s a tag with bright signal, with low background and good photostability. For small cell components like organelles, the tag must be cell-permeable and small enough to not interfere with normal cellular processes such as transcription and metabolism.
Significant advances have been made in fluorescent tags in the past two decades. Here we look at several papers noting these advances.
The first monoclonal antibody (mAb) was produced in a lab 1975, and the first therapeutic mAb was introduced in the United States to prevent kidney transplant rejection in 1986. The first mAb used in cancer treatment the anti-CD20 mAb, rituximab, was used to treat non-Hodgkin’s lymphoma and chronic lymphocytic leukemia. Today therapeutic mAbs have become a mainstay of cancer, autoimmune disease, and metabolic disease therapies and include HERCEPTIN® used to treat certain forms of breast cancer, Prolia used to treat bone loss in post-menopausal women, and Stelara used to treat autoimmune diseases like psoriatic arthritis and severe Crohn disease, among many others. Therapeutic mAbs bind targets with high specificity and affinity and they can recruit effector cells to drive target elimination through mechanisms such as antibody-dependent cellular cytotoxicity (ADCC) or antibody-dependent cellular phagocytosis (ADCP), making them highly specific, effective therapies.
Methylglyoxal is responsible for post translational protein modifications, that result in advanced glycation endproducts (AGEs), which are associated with aging and disease.
Post-translational modifications (PTM) of proteins are essential for the function of many proteins, but aberrant modification of protein residues also can interfere with protein function. PTMs occur in two ways. Proteins may be modified through the activity of enzymes such as kinases, phosphorylases, glycosylases and others that add or remove specific chemical moieties to amino acid residues. PTMs can also result from non-enzymatic reaction between electrophilic compounds and nucleophilic arginine and lysine residues within a protein. Metabolites and metabolic by products produced during glycolysis, especially glyoxal and methylglyoxal (MGO), are examples of such electrophilic compounds. These compounds can react with arginine and lysine to produce advanced glycation end products (AGEs), which are biomarkers associated with aging and degenerative diseases such as Alzheimer disease, diabetes and others. MGO is also elevated in tumors that have switched from oxidative phosphorylation to glycolysis as their main energy production pathway.
Only limited information is available about site-specific MGO PTMs in mammal cells, and most studies have focused on measuring the amount of MGO modifications in a treatment scenario compared to a control. Donnellan and colleagues recently published work to identify specific MGO protein modifications. They used a “bottom-up” proteomic analysis of WIL2-NS B lymphoblastoid whole-cell lysates to identify specific MGO-modified proteins. In particular, the group was looking for modifications in proteins that might explain how MGO activity contributes to aneuploidy.
For the study, 100µg of cellular protein extract was reduced with dithiothreitol and then alkylated with chloroacetamide. The sample was diluted to reduce urea concentration. Trypsin Gold was added and samples were digested for 8 hours at 37°C. Digestion was terminated by adding formic acid. For ProAlanase digestion, 20µg of protein was reduced, alkylated and diluted to reduce urea concentration before adding digesting with ProAlanase for 4 hours at 37°C.
Are you looking for proteases to use in your research? Explore our portfolio of proteases today.
The authors identified 519 MGO-modified proteins. Most of the modifications were identified in the trypsin digestion reactions; however, ProAlanase digestion increased the number of MGO modifications identified by approximately 25% (with less than 4% of the modification sites being detected in both the ProAlanase and trypsin digestion reactions. The authors suggest that ProAlanase increased sequence coverage to reveal sites not detected in the trypsin digestions. Therefore, they conclude that ProAlanase can be used along with trypsin digestion to increase the identification of MGO modifications.
ProAlanase can be used along with trypsin digestion to increase the identification of MGO modifications.
MGO-modified proteins from the WIL2-NS whole cell lysates included proteins involved in glycolysis, translation initiation, protein folding, mRNA splicing, cell-to-cell adhesion, heat response, nucleosome assembly, protein SUMOylation and the G2/M cell cycle transition. More work to further characterize the sites of these modifications and their potential effects on the function of the modified proteins is ongoing.
Donnelian, L et al. (2022) Proteomic Analysis of Methylglyoxal Modifications Reveals Susceptibility of Glycolytic Enzymes to Dicarbonyl Stress Int. J. Mol. Sci.23(7), 3689 doi.org/10.3390/ijms23073689
Biologic therapeutics such as monoclonal antibodies and biosimilars are complex proteins that are susceptible to post-translational modifications (PTMs). These chemical modifications can affect the performance and activity of the biologic, potentially resulting in decreased potency and increased immunogenicity. Such modifications include glycosylation, deamidation, oxidation and disulfide bond shuffling. These PTMs can be signs of protein degradation, manufacturing issues or improper storage. Several of these modifications are well characterized, and methods exist for detecting them during biologic manufacture. However, disulfide shuffling is not particularly well characterized for biologics, and no methods exist to easily detect and quantify disulfide bond shuffling in biologics.
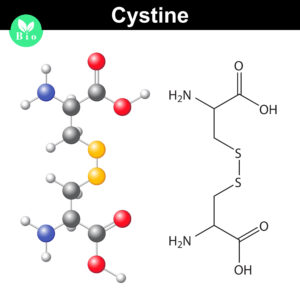
Disulfide bonds are important for protein conformation and function
Normally the cysteines in a protein will pair with a predictable or “normal” partner residue either within a polypeptide chain or between two polypeptide chains when they form disulfide bonds. These normal disulfide bonds are important for final protein conformation and stability. Indeed, disulfide bonds are considered an important quality indicator for biologics.
In a recently published study, Coghlan and colleagues designed a semi-automated method for characterizing disulfide bond shuffling on two IgG1 biologics: rituximab (originator drug Rituxan® and biosimilar Acellbia®) and bevacizumab (originator Avastin® and biosimilar Avegra®).
XWe use cookies and similar technologies to make our website work, run analytics, improve our website, and show you personalized content and advertising. Some of these cookies are essential for our website to work. For others, we won’t set them unless you accept them. To learn more about our approach to Privacy we invite you to Read More
By clicking “Accept All”, you consent to the use of ALL the cookies. However you may visit Cookie Settings to provide a controlled consent.
We use cookies and similar technologies to make our website work, run analytics, improve our website, and show you personalized content and advertising. Some of these cookies are essential for our website to work. For others, we won’t set them unless you accept them. To find out more about cookies and how to manage cookies, read our Cookie Policy.
If you are located in the EEA, the United Kingdom, or Switzerland, you can change your settings at any time by clicking Manage Cookie Consent in the footer of our website.
Necessary cookies are absolutely essential for the website to function properly. These cookies ensure basic functionalities and security features of the website, anonymously.
Cookie
Duration
Description
cookielawinfo-checbox-analytics
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Analytics".
cookielawinfo-checbox-functional
11 months
The cookie is set by GDPR cookie consent to record the user consent for the cookies in the category "Functional".
cookielawinfo-checbox-others
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Other.
cookielawinfo-checkbox-advertisement
1 year
The cookie is set by GDPR cookie consent to record the user consent for the cookies in the category "Advertisement".
cookielawinfo-checkbox-necessary
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookies is used to store the user consent for the cookies in the category "Necessary".
cookielawinfo-checkbox-performance
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Performance".
gdpr_status
6 months 2 days
This cookie is set by the provider Media.net. This cookie is used to check the status whether the user has accepted the cookie consent box. It also helps in not showing the cookie consent box upon re-entry to the website.
lang
This cookie is used to store the language preferences of a user to serve up content in that stored language the next time user visit the website.
viewed_cookie_policy
11 months
The cookie is set by the GDPR Cookie Consent plugin and is used to store whether or not user has consented to the use of cookies. It does not store any personal data.
Analytical cookies are used to understand how visitors interact with the website. These cookies help provide information on metrics the number of visitors, bounce rate, traffic source, etc.
Cookie
Duration
Description
SC_ANALYTICS_GLOBAL_COOKIE
10 years
This cookie is associated with Sitecore content and personalization. This cookie is used to identify the repeat visit from a single user. Sitecore will send a persistent session cookie to the web client.
vuid
2 years
This domain of this cookie is owned by Vimeo. This cookie is used by vimeo to collect tracking information. It sets a unique ID to embed videos to the website.
WMF-Last-Access
1 month 18 hours 24 minutes
This cookie is used to calculate unique devices accessing the website.
_ga
2 years
This cookie is installed by Google Analytics. The cookie is used to calculate visitor, session, campaign data and keep track of site usage for the site's analytics report. The cookies store information anonymously and assign a randomly generated number to identify unique visitors.
_gid
1 day
This cookie is installed by Google Analytics. The cookie is used to store information of how visitors use a website and helps in creating an analytics report of how the website is doing. The data collected including the number visitors, the source where they have come from, and the pages visted in an anonymous form.
Advertisement cookies are used to provide visitors with relevant ads and marketing campaigns. These cookies track visitors across websites and collect information to provide customized ads.
Cookie
Duration
Description
IDE
1 year 24 days
Used by Google DoubleClick and stores information about how the user uses the website and any other advertisement before visiting the website. This is used to present users with ads that are relevant to them according to the user profile.
test_cookie
15 minutes
This cookie is set by doubleclick.net. The purpose of the cookie is to determine if the user's browser supports cookies.
VISITOR_INFO1_LIVE
5 months 27 days
This cookie is set by Youtube. Used to track the information of the embedded YouTube videos on a website.
Performance cookies are used to understand and analyze the key performance indexes of the website which helps in delivering a better user experience for the visitors.
Cookie
Duration
Description
YSC
session
This cookies is set by Youtube and is used to track the views of embedded videos.
_gat_UA-62336821-1
1 minute
This is a pattern type cookie set by Google Analytics, where the pattern element on the name contains the unique identity number of the account or website it relates to. It appears to be a variation of the _gat cookie which is used to limit the amount of data recorded by Google on high traffic volume websites.