Post-translational modifications of proteins are critical for proper protein function. Modifications such as phosphorylation/dephosphorylation can act as switches that activate or inactivate proteins in signaling cascades. The addition of specific sugars to membrane proteins on cells are critical for recognition, interaction with the extracellular matrix and other activities. While we know volumes about some types of protein modifications, ADP-ribosylation on aspartate and glutamate residues has been more difficult to study because of the chemical instability of these ester-linked modifications.
Matić Lab (Eduardo José Longarini and Ivan Matić) recently published a study that explored mono-ADP-ribosylation (ADPr) on aspartate and glutamate residues by the protein PARP1 and its potential reversal by PARG. PARP1 and PARG signaling are central to DNA repair and apoptosis pathways, making them potentially powerful therapeutic targets in cancer or neurodegenerative diseases in which DNA repair processes are often disrupted.
Biologic therapeutics such as monoclonal antibodies and biosimilars are complex proteins that are susceptible to post-translational modifications (PTMs). These chemical modifications can affect the performance and activity of the biologic, potentially resulting in decreased potency and increased immunogenicity. Such modifications include glycosylation, deamidation, oxidation and disulfide bond shuffling. These PTMs can be signs of protein degradation, manufacturing issues or improper storage. Several of these modifications are well characterized, and methods exist for detecting them during biologic manufacture. However, disulfide shuffling is not particularly well characterized for biologics, and no methods exist to easily detect and quantify disulfide bond shuffling in biologics.
Disulfide bonds are important for protein conformation and function
Normally the cysteines in a protein will pair with a predictable or “normal” partner residue either within a polypeptide chain or between two polypeptide chains when they form disulfide bonds. These normal disulfide bonds are important for final protein conformation and stability. Indeed, disulfide bonds are considered an important quality indicator for biologics.
In a recently published study, Coghlan and colleagues designed a semi-automated method for characterizing disulfide bond shuffling on two IgG1 biologics: rituximab (originator drug Rituxan® and biosimilar Acellbia®) and bevacizumab (originator Avastin® and biosimilar Avegra®).
The spike protein of the SARS-CoV-2 virus is a very commonly researched target in COVID-19 vaccine and therapeutic studies because it is an integral part of host cell entry through interactions between the S1 subunit of the spike protein with the ACE2 protein on the target cell surface. Viral proteins important in host cell entry are typically highly glycosylated. Looking at the sequence of the SARS-CoV-2 virus, researchers predict that the spike protein is highly glycosylated. In a recent study, researchers conducted a glycosylation analysis of SARS-CoV-2 proteins using mass spec analysis to determine the N-glycosylation profile of the subunits that make up the spike protein.
Glycans assist in protein folding and help the virus avoid immune recognition by the host. Glycosylation can also have an impact on the antigenicity of the virus, as well as potential effects on vaccine safety and efficacy. Mass spectrometry is widely used for viral characterization studies of influenza viruses. Specifically, mass spec has been used to study influenza protein glycosylation, antigen quantification, and determination of vaccine potency.
The use of mass spectrometry for the characterization of individual or complex protein samples continues to be one of the fastest growing fields in the life science market.
Bottom-up proteomics is the traditional approach to address these questions. Optimization of each the individual steps (e.g. sample prep, digestion and instrument performance) is critical to the overall success of the entire experiment.
To address issues that may arise in your experimental design, Promega has developed unique tools and complementary webinars to help you along the way.
Here you can find a summary of individual webinars for the following topics:
It’s time to analyze your protein and you are trying to decide where to begin. You are asking questions like: Which protease do I choose? How much enzyme should I use in my digest? How long should I perform my digest?
Unfortunately, there is no one-size fits all answer to this type of question other than… “well it depends.” All protease digests will be a balance between denaturing the protein sample to allow access to cleavage sites, optimizing conditions for the protease to function, and compatibility with your workflow and downstream applications. We provide general guidelines that work for most samples, but frequently you will need to optimize the conditions need for your specific sample and application.
Here, I use the example of a trypsin digest for downstream mass spectrometry to highlight key questions to ask and factors that can be optimized for any digest.
Asp-N, Sequencing Grade, is an endoproteinase that hydrolyzes peptide bonds on the N-terminal side of aspartic and cysteic acid residues: Asp and Cys. Asp-N activity is optimal in the pH range of 4.0–9.0. This sequencing grade enzyme can be used alone or in combination with trypsin or other proteases to produce protein digests for peptide mapping applications or protein identification by peptide mass fingerprinting or MS/MS spectral matching. It is suitable for in-solution or in-gel digestion reactions.
The following references illustrate the use of Asp-N in recent publications:
Protein sequence coverage
Jakobsson, M et al. (2013) Identification and characterization of a novel Human Methyltransferase modulating Hsp70 protein function through lysine methylation. J. Biol. Chem. 288, 27752–63.
Carroll, J. et. al. (2013) Post-translational modifications near the quinone binding site of mammalian complex I. J. Biol. Chem. 288, 24799–08.
Glycoprotein analysis
Siguier, B. et al. (2014) First structural insights into α-L-Arabinofuranosidases from the two GH62 Glycoside hydrolase subfamilies. J. Biol. Chem. 289, 5261–73.
Vakhrushev, S. et al. (2013) Enhanced mass spectrometric mapping of the human GalNAc-type O-glycoproteome with SimpleCells. Mol. Cell. Prot.12, 932–44.
Berk, J. et al. (2013) . O-Linked β-N- Acetylglucosamine (O-GlcNAc) Regulates emerin binding to autointegration Factor (BAF) in a chromatin and Lamin B-enriched “Niche”. J. Biol. Chem. 288, 30192–09.
Phosphoprotein analysis
Roux, P. and Thibault, P. (2013) The Coming of Age of phosphoproteomics –from Large Data sets to Inference of protein Functions. Mol. Cell. Prot.12, 3453–64.
Are you looking for proteases to use in your research? Explore our portfolio of proteases today.
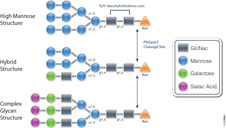
PNGase F (Cat.# V4831) is a recombinant glycosidase cloned from Elizabethkingia meningoseptica and overexpressed in E. coli, with a molecular weight of 36kD.
PNGase F catalyzes the cleavage of N-linked oligosaccharides between the innermost GlcNAc and asparagine residues of high mannose, hybrid, and complex oligosaccharides from N-linked glycoproteins. PNGase F will not remove oligosaccharides containing alpha-(1,3)-linked core fucose, commonly found on plant glycoproteins.
Applications Determining whether a protein is in fact glycosylated is the initial step in glycoprotein analysis. Polyacrylamide gel electrophoresis in the presence of sodium dodecyl sulfate (SDS-PAGE) has become the method of choice as the final step prior to mass spec analysis. Glycosylated proteins often migrate as diffused bands by SDS-PAGE. A marked decrease in band width and change in migration position after treatment with PNGase F is considered evidence of N-linked glycosylation.
Gel based data are often correlated with information obtained from mass spec analysis. Asn-linked type glycans can be cleaved enzymatically by PNGase F yielding intact oligosaccharides and a slightly modified protein in which Asn residues at the site of de-N-glycosylation are converted to Asp, by converting the previously carbohydrate-linked asparagine into an aspartic acid, a monoisotopic mass shift of 0.9840Da is observed. The deglycosylated peptides are then analyzed by tandem mass spectrometry (MS/MS), and software algorithms are used to correlate the experimental fragmentation spectra with theoretical tandem mass spectra generated from peptides in a protein database.
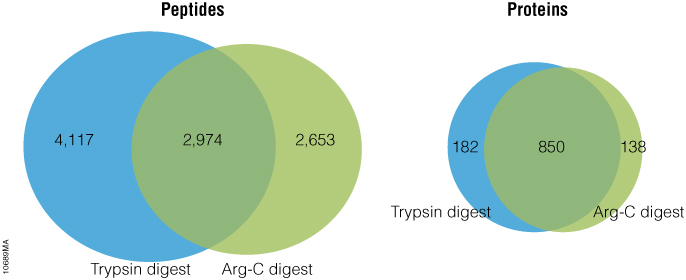
Arg-C (clostripain), Sequencing Grade (Cat.# V1881), is a specific endoproteinase isolated from the soil bacterium Clostridium histolyticum. It preferentially cleaves at the C-terminal side of arginine (R) residues. Unlike trypsin, Arg-C efficiently cleaves arginine sites followed by proline (P). This difference is important because every twentieth arginine is followed by proline. To illustrate this benefit, Arg-C was evaluated for protein analysis in two different experiments. In the first experiment, we studied the use of Arg-C for proteomic analysis. Yeast provides an excellent model proteome because its genome is well annotated. Yeast extract was digested in two parallel reactions, using trypsin in the first reaction and Arg-C in the second, using a conventional protocol consistent with LC-MS/MS analysis. As expected the trypsin digestion resulted in a high number of peptide and protein identifications (Figure 1). However, many peptides remained elusive. The parallel Arg-C digestion complemented the trypsin digestion by recovering an additional 2,653 peptides and providing a 37.4% increase in the number of identified peptides. Digesting with Arg-C also resulted in an increase in the number of identified proteins. In fact, 138 new proteins were identified in Arg-C digest compared to the parallel trypsin digest, offering a 13.4% increase in the overall number of identified proteins.
Figure 1. Side-by-side analysis of trypsin-digested and Arg-C digested yeast proteins.
Are you looking for proteases to use in your research? Explore our portfolio of proteases today.
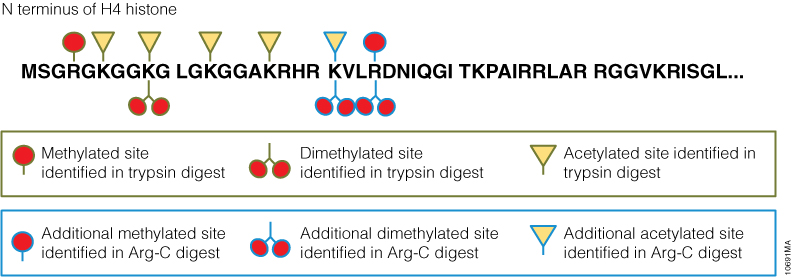
In a second experiment, the ability of Arg-C to analyze individual proteins was analyzed, selecting human histone H4 as a model protein. Like other histones, this protein is heavily modified post translational modifications (PTMs) that alter histone structure and regulate interaction with transcription factors. As a result, histone PTMs are implicated in gene regulation and associated with multiple disorders. Technical challenges, however, impede histone PTM analysis. Histone PTMs are complex and some, such as acetylation and methylation, prevent trypsin digestion, as shown by our data. In this experiment, trypsin digestion of histone H4 identified several PTMs (Figure 2). However, certain PTMs were missing. By digesting histone H4 with Arg-C, we were able to identify the missing PTMs including mono-, dimethylated and acetylated lysine and arginine residues. We speculate that the PTMs in human histone H4, which modified arginine and lysine residues, rendered trypsin unsuitable for preparing the corresponding histone regions for mass spectrometry. The problem was rectified by replacing trypsin with Arg-C.
Figure 2. Identification of histone h4 PTMs after Arg-C digestion.
One of the approaches to identify proteins by mass spectrometry includes the separation of proteins by gel electrophoresis or liquid chromatography. Subsequently the proteins are cleaved with sequence-specific endoproteases. Following digestion the generated peptides are investigated by determination of molecular masses or specific sequence. For protein identification the experimentally obtained masses/sequences are compared with theoretical masses/sequences compiled in various databases.
Are you looking for proteases to use in your research? Explore our portfolio of proteases today.
Nonspecific proteases such as pepsin, proteinase K, elastase and thermolysin can offer an alternative to traditional sequence-specific proteases for certain applications. The following references illustrate the use of nonspecific proteinases for the mass spec analysis of proteins:
XWe use cookies and similar technologies to make our website work, run analytics, improve our website, and show you personalized content and advertising. Some of these cookies are essential for our website to work. For others, we won’t set them unless you accept them. To learn more about our approach to Privacy we invite you to Read More
By clicking “Accept All”, you consent to the use of ALL the cookies. However you may visit Cookie Settings to provide a controlled consent.
We use cookies and similar technologies to make our website work, run analytics, improve our website, and show you personalized content and advertising. Some of these cookies are essential for our website to work. For others, we won’t set them unless you accept them. To find out more about cookies and how to manage cookies, read our Cookie Policy.
If you are located in the EEA, the United Kingdom, or Switzerland, you can change your settings at any time by clicking Manage Cookie Consent in the footer of our website.
Necessary cookies are absolutely essential for the website to function properly. These cookies ensure basic functionalities and security features of the website, anonymously.
Cookie
Duration
Description
cookielawinfo-checbox-analytics
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Analytics".
cookielawinfo-checbox-functional
11 months
The cookie is set by GDPR cookie consent to record the user consent for the cookies in the category "Functional".
cookielawinfo-checbox-others
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Other.
cookielawinfo-checkbox-advertisement
1 year
The cookie is set by GDPR cookie consent to record the user consent for the cookies in the category "Advertisement".
cookielawinfo-checkbox-necessary
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookies is used to store the user consent for the cookies in the category "Necessary".
cookielawinfo-checkbox-performance
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Performance".
gdpr_status
6 months 2 days
This cookie is set by the provider Media.net. This cookie is used to check the status whether the user has accepted the cookie consent box. It also helps in not showing the cookie consent box upon re-entry to the website.
lang
This cookie is used to store the language preferences of a user to serve up content in that stored language the next time user visit the website.
viewed_cookie_policy
11 months
The cookie is set by the GDPR Cookie Consent plugin and is used to store whether or not user has consented to the use of cookies. It does not store any personal data.
Analytical cookies are used to understand how visitors interact with the website. These cookies help provide information on metrics the number of visitors, bounce rate, traffic source, etc.
Cookie
Duration
Description
SC_ANALYTICS_GLOBAL_COOKIE
10 years
This cookie is associated with Sitecore content and personalization. This cookie is used to identify the repeat visit from a single user. Sitecore will send a persistent session cookie to the web client.
vuid
2 years
This domain of this cookie is owned by Vimeo. This cookie is used by vimeo to collect tracking information. It sets a unique ID to embed videos to the website.
WMF-Last-Access
1 month 18 hours 24 minutes
This cookie is used to calculate unique devices accessing the website.
_ga
2 years
This cookie is installed by Google Analytics. The cookie is used to calculate visitor, session, campaign data and keep track of site usage for the site's analytics report. The cookies store information anonymously and assign a randomly generated number to identify unique visitors.
_gid
1 day
This cookie is installed by Google Analytics. The cookie is used to store information of how visitors use a website and helps in creating an analytics report of how the website is doing. The data collected including the number visitors, the source where they have come from, and the pages visted in an anonymous form.
Advertisement cookies are used to provide visitors with relevant ads and marketing campaigns. These cookies track visitors across websites and collect information to provide customized ads.
Cookie
Duration
Description
IDE
1 year 24 days
Used by Google DoubleClick and stores information about how the user uses the website and any other advertisement before visiting the website. This is used to present users with ads that are relevant to them according to the user profile.
test_cookie
15 minutes
This cookie is set by doubleclick.net. The purpose of the cookie is to determine if the user's browser supports cookies.
VISITOR_INFO1_LIVE
5 months 27 days
This cookie is set by Youtube. Used to track the information of the embedded YouTube videos on a website.
Performance cookies are used to understand and analyze the key performance indexes of the website which helps in delivering a better user experience for the visitors.
Cookie
Duration
Description
YSC
session
This cookies is set by Youtube and is used to track the views of embedded videos.
_gat_UA-62336821-1
1 minute
This is a pattern type cookie set by Google Analytics, where the pattern element on the name contains the unique identity number of the account or website it relates to. It appears to be a variation of the _gat cookie which is used to limit the amount of data recorded by Google on high traffic volume websites.