Protein degrader research has yielded its first approved therapeutic for specific breast cancer patients: Vepdegestrant received FDA approval on May 1, 2026 (1). Vepdegestrant is an oral PROteolysis TArgeting Chimera (PROTAC) that targets the estrogen receptor for degradation in breast cancer patients with ESR1-mutated ER+/HER2– advanced breast cancer (2) produced by Arvinas, Inc. in collaboration with Pfizer Inc.
A Different Kind of Drug Development
Targeted protein degraders (or PROTACs) have opened new possibilities in drug discovery research. Instead of inhibiting protein function or interaction, degraders cause the removal of the target protein itself. Traditional small molecule drugs work by binding a protein to inhibit it or block function, and they must remain bound to work. That means that the target protein should be well-characterized in terms of binding and activity sites, and the drug must bind specifically only to the target protein. In contrast, degraders only need to bind long enough to recruit cellular protein degradation machinery to the target protein, and the method does not rely on an accessible and specific binding site on the target protein. Once degradation occurs, the degrader is released and can engage with the next target.
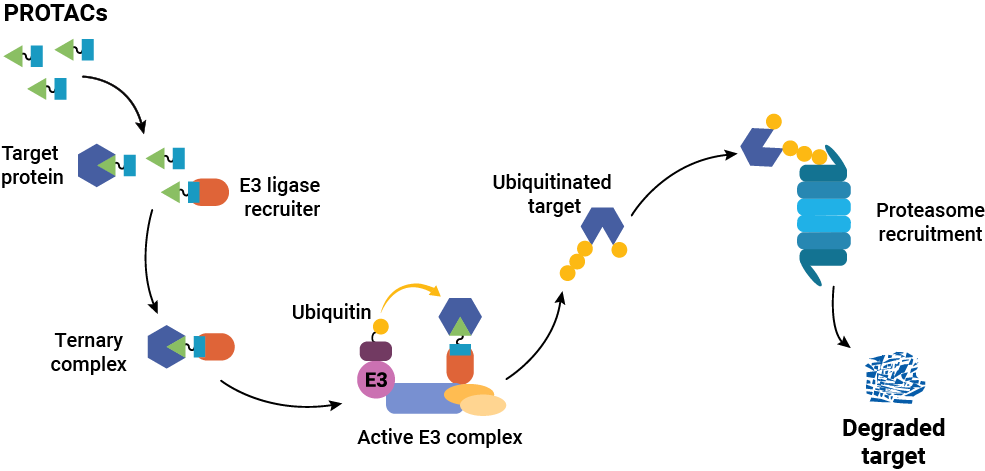
The approval of vepdegestrant is a landmark moment for the entire TPD and induced proximity field, demonstrating that it is possible to rationally design molecules whose pharmacology is categorically distinct from traditional drugs, relying on a catalytic rather than occupancy-driven mechanism of action. More importantly, this translates to meaningful clinical outcomes in patients. —Dr. Kristin Riching, Promega R&D Scientist
The first peptide-based PROTAC was described in 2001 in the laboratories of Craig Crews and Ray Deshaies (3), but translating the concept into orally bioavailable, clinically viable molecules took nearly two decades, using tools that did not exist when the field began. More than 40 PROTAC degraders have now entered clinical trials (4), with vepdegestrant the most advanced, supported by Phase 3 data from the VERITAC-2 trial demonstrating statistically significant improvement in progression-free survival in ESR1-mutant patients. That progress required solving a measurement problem as much as a chemistry one: how do you quantify target protein degradation at endogenous levels, with enough sensitivity and throughput to drive a screening campaign? CRISPR-engineered protein tagging combined with the small bioluminescent reporter tag, HiBiT solved that problem, providing a sensitive, HTS-compatible readout of endogenous target levels without relying on laborious, artifact-prone western blots. Critically, HiBiT also enabled researchers to watch target protein degradation unfold in real time in living cells.
“Seeing it happen in real time, frankly, may have been what convinced many people that the modality had genuine merit.”
—Dr. Kristin Riching
Developing a PROTAC is not like developing a traditional inhibitor. Success requires successful completion of a complex cascade of cellular events: the molecule must enter the cell, engage the target protein and the E3 ligase simultaneously, form a productive ternary complex in the right geometry, trigger ubiquitination, and drive proteasomal degradation, all while competing with cellular noise that can blunt each step. “PROTACs are large molecules, so they are often not very permeable,” Riching explains. “They also need to simultaneously engage both the target and the E3 ligase machinery, but they need to do so in a productive geometry that leads to ubiquitination, which is not easily predicted. In cells, many compounding factors can limit activity, making it difficult to identify which parameters most need improvement. Event-driven modalities like PROTACs rely on robust tools to tease apart each mechanistic step to aid SAR optimization.”
Getting that data means adopting a screening framework built around mechanistic understanding of the full degradation cascade from the earliest stages of optimization, while preserving the native biology and the stoichiometric relationships that govern degradation efficiency. It also means going beyond endpoint measurements. Knowing whether a target is degraded is a starting point; knowing how fast, how completely, and how durably it degrades is what distinguishes a development candidate from a dead end. Riching’s research has shown that different proteins in the same family can respond to the same PROTAC with dramatically different kinetic profiles (5,6), which is a distinction that endpoint assays cannot capture, and one that can determine which compounds are worth advancing.
What’s Next after Vepdegestrant?
The approval of Vepdegestrant validates more than just a single drug, it validates the PROTAC drug category and the tools and methods that enabled it. For researchers working on next-generation degraders, the signal is clear: the modality works. Now the question is how far we can push it.
Riching points to E3 ligase diversity as the field’s most pressing unresolved problem. “The greatest challenge will be expanding beyond the two E3 ligases — CRBN and VHL — that have driven most PROTAC progress to date,” she says. “We don’t yet fully understand the scope of targets accessible through these ligases, but it stands to reason that additional ligases will be necessary to unlock a larger portion of the degradable proteome. Their broad distribution also limits opportunity for tissue-selective targeting. Developing the tools and chemistry to recruit a wider repertoire of E3 ligases remains one of the most important unsolved problems the field faces.”
Beyond ligase diversity, the field is expanding its conception of what a degrader can be. Molecular glues, LYTACs, and other induced proximity strategies are broadening the range of accessible targets — including extracellular and membrane-bound proteins that sit outside the reach of classical PROTACs. Each new modality brings its own characterization challenges, and the same principle holds: understanding mechanism at the cellular level, early and rigorously, is what separates the compounds worth advancing from those that look promising in a tube.
The approval of vepdegestrant is a landmark. But researchers working in this space know it is a beginning as much as it is a culmination — proof that the approach is sound, and a starting line for everything that follows.
Read more about Innovative Imaging Solutions for Targeted Protein Degradation on our website.
Literature Cited
- Arvinas, Inc. Arvinas Announces FDA Approval of VEPPANU (vepdegestrant) for the Treatment of ESR1m, ER+/HER2– Advanced Breast Cancer Accessed: May 5, 2026
- Arvinas, Inc. (2025) Arvinas Announces FDA Acceptance of the New Drug Application for Vepdegestrant for the Treatment of ESR1m, ER+/HER2– Advanced Breast Cancer. August 8. Accessed: April 27, 2026.
- Sakamoto, K.M. et al. (2001) Protacs: Chimeric Molecules that Target Proteins to the Skp1-Cullin-F Box Complex for Ubiquitination and Degradation. Proc. Natl. Acad. Science USA 98, 8554-–9. Accessed: May 4, 2026.
- Chen, S. (2026) Protein Degraders (PROTACS & Molecular Glues) in 2026: The Emergining Challenge to Traditional Drug Development Accessed: May 5, 2026
- Riching, K.M et al. (2018) Quantitative Live-Cell Kinetic Degradation and Mechanistic Profiling of PROTAC Mode of Action. ACS Chem. Biol. 13, 2758–70. Accessed: May 4, 2026
- Riching, K.M. et al. (2022) The Importance of Cellular Degradation Kinetics for Understanding Mechanisms in Targeted Protein Degradation. Chem. Soc. Rev. 51, 6210–6221.
This article was written with AI assistance.