Our cells have evolved multiple mechanisms for “taking out the trash”—breaking down and disposing of cellular components that are defective, damaged or no longer required. Within a cell, these processes are balanced by the synthesis of new components, so that DNA, RNA and proteins are constantly undergoing turnover.
Proteins are degraded by two major components of the cellular machinery. The discovery of the lysosome in the mid-1950s provided considerable insight into the first of these degradation mechanisms for extracellular and cytosolic proteins. Over the next several decades, details of a second protein degradation mechanism emerged: the ubiquitin-proteasome system (UPS). Ubiquitin is a small, highly conserved polypeptide that is used to selectively tag proteins for degradation within the cell. Multiple ubiquitin tags are generally attached to a single targeted protein. This ill-fated, ubiquitinated protein is then recognized by the proteasome, a large protein complex with proteolytic activity. Ubiquitination is a multistep process, involving several specialized enzymes. The final step in the process is mediated by a family of ubiquitin ligases, known as E3.
The importance of the UPS extends beyond its function as a cellular garbage disposal system. Regulated proteolysis plays a role in a wide range of cellular processes, from the cell cycle and cell division to the modulation of metabolic pathways.
Traditionally, small-molecule drug discovery efforts have been focused around drugs that can specifically bind to and inhibit the activity of a target protein. However, there’s a problem with that approach: up to 85% of the proteins within the cell are “undruggable”, meaning that they lack accessible binding sites for small molecules. Naturally, this challenge has spurred the drive to find new and creative ways to develop small-molecule drugs that overcome the limitation of binding to an active site on the target protein.
Our increased understanding of the UPS offers an attractive alternative: what if you could simply tag an unwanted protein for degradation, such as the misfolded or damaged protein aggregates involved in a variety of neurodegenerative diseases? Conversely, many cancer pathways rely on degrading tumor suppressor proteins and cell cycle checkpoint inhibitors; in these scenarios, small-molecule inhibitors of key UPS components could have significant therapeutic value. The “miracle molecules” that can make such dreams a reality belong to two classes of compounds: proteolysis-targeting chimeras, or PROTACs, and molecular glues.
PROTACS and Molecular Glues
As their name implies, PROTACs are hybrid, bifunctional molecules. One moiety attaches to the target protein, and the other binds to a component of the ubiquitin E3 ligase complex. The two parts of a PROTAC are connected by a linker. Ideally, a PROTAC would engage the target protein and E3 ligase component simultaneously to form a “ternary complex”. This complex results in ubiquitination of the target protein and subsequent transport to the proteasome for digestion. While early PROTACs were small peptides, subsequent advances in PROTAC discovery and development were driven by small-molecule drugs (reviewed by Chamberlain and Hamann, 2019).
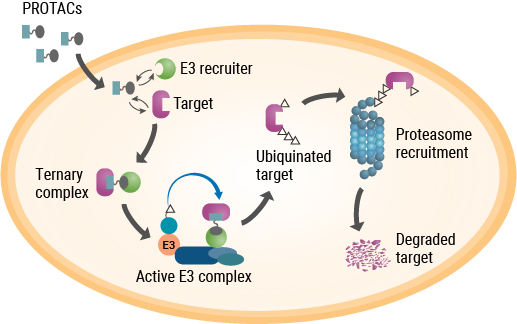
Unlike PROTACs, molecular glues are not bifunctional molecules. Rather, they facilitate the interaction between two proteins that would otherwise have low affinity for each other—in this case, two components of the UPS. A drug that spurred research into the mechanism of molecular glues was one with a notorious past—thalidomide. Withdrawn from the market due to its potent teratogenic effects, thalidomide and several analogs returned to clinical development due to their efficacy in treating complications of leprosy and multiple myeloma, although the precise mechanism of action remained unknown. As it turned out, thalidomide binds to (and was affinity-purified with) two proteins—cereblon and DNA damage binding protein 1 (DDB1)—that are components of the ubiquitin E3 ligase complex. Further studies showed that thalidomide binding to cereblon facilitates protein-protein interactions that ultimately target several transcription factors for degradation (Chamberlain and Hamann, 2019).
Studying Protein Degradation in Live Cells
Clearly, unraveling the mechanistic pathways involved in targeted protein degradation has opened up new opportunities for drug discovery and development. These efforts require sensitive methods to detect endogenous proteins and measure their degradation in response to the compounds being evaluated. Ideally, such methods would monitor degradation in real time and be amenable to high-throughput formats required to screen large compound libraries. Promega has developed methods, based on NanoLuc® technology, to interrogate various stages of the protein degradation pathway (reviewed by Daniels et al., 2019). These methods will advance the understanding of targeted protein degradation and provide effective tools to accelerate the pace of drug development within this rapidly growing field.
Learn about the latest tools for studying protein degradation at the North American Protein Degradation Congress in San Diego, CA, February 4–6, 2020. For more information, including protocols and applications, visit the Promega targeted protein degradation resource page.
Related Posts
Latest posts by Ken Doyle (see all)
- Will Artificial Intelligence (AI) Transform the Future of Life Science Research? - February 1, 2024
- RAF Inhibitors: Quantifying Drug-Target Occupancy at Active RAS-RAF Complexes in Live Cells - September 5, 2023
- Synthetic Biology: Minimal Cell, Maximal Opportunity - July 25, 2023