This post is written by Kai Hillman, PhD, Promega Corporation.
Every day, scientists push the boundaries of what’s possible with monoclonal antibodies (mAbs)—from targeting cancer cells to calming autoimmune-driven inflammation. These therapies rely not only on binding but on engineering the desired immune response. The suite of Promega Fc Effector Assays helps you understand these interactions from receptor binding and function, through bridging studies. With consistency, sensitivity, and scalability, these assays support teams from early discovery through lot release.
This article draws on real-world publications and product insights to show how Promega assays are powering next-generation immunotherapies—and redefining how we measure immune engagement.
Approximately 30 million years ago, a retrovirus integrated into the germline of a common ancestor of baboons, gorillas, chimpanzees and humans. That endogenous retrovirus, now known as gammaretrovirus human endogenous retrovirus 1 (HERV-1), may provide clues about the aberrant regulation of gene transcription that enables tumor cells to grow and survive.
Understanding the Mechanism Behind Cancer Gene Expression
Scientists have long described the striking differences in gene expression, signaling activity and metabolism between cancer cells and normal cells, but the underlying mechanisms that cause these differences are not fully understood. In a recent Science Advancesarticle, published by Ivancevic et al., researchers from the University of Colorado, Boulder; the University of Colorado Anschutz Medical Campus, and the University of Colorado School of Medicine report their efforts to identify endogenous retrovirus elements that might be part of the answer to the complex question of what biological events are responsible for the changes in gene expression in cancer cells.
The researchers hypothesized that transposable elements (TEs), specifically those associated with endogenous retroviruses could be involved in cancer-specific gene regulation. Endogenous retroviruses (ERVs) are the remnants of ancient retroviral infections that have integrated into the germline of the host.
Identifying Endogenous Retrovirus Elements That Affect Cancer Gene Expression
This post was contributed by guest blogger, Scott Messenger, Technical Support Scientist 2 at Promega Corporation.
It’s always an exciting time in the lab when you find a new assay to answer an important research question. Once you get your hands on the assay, it is always good to confirm it will work for your experimental setup. Repeating the control experiment shown in the technical manual is a great way to test the assay in your hands.
After running that first experiment of your assay, it looks pretty good. The trends of control and treatment are consistent. Time to get on with the experiments…but wait—the RLUs (Relative Light Units) are two orders of magnitude lower than the example data! I can’t show this data to my colleagues; it doesn’t match. What did I do wrong?
This is a concern that we in Technical Services hear frequently. The concern is real, and I had this same thought when doing some of my first experiments using luminescence. When a question like this comes in, a Technical Service Scientist will make sure the experiment was performed as we described, and in most cases it is. We then start talking about RLUs (Relative Light Units).
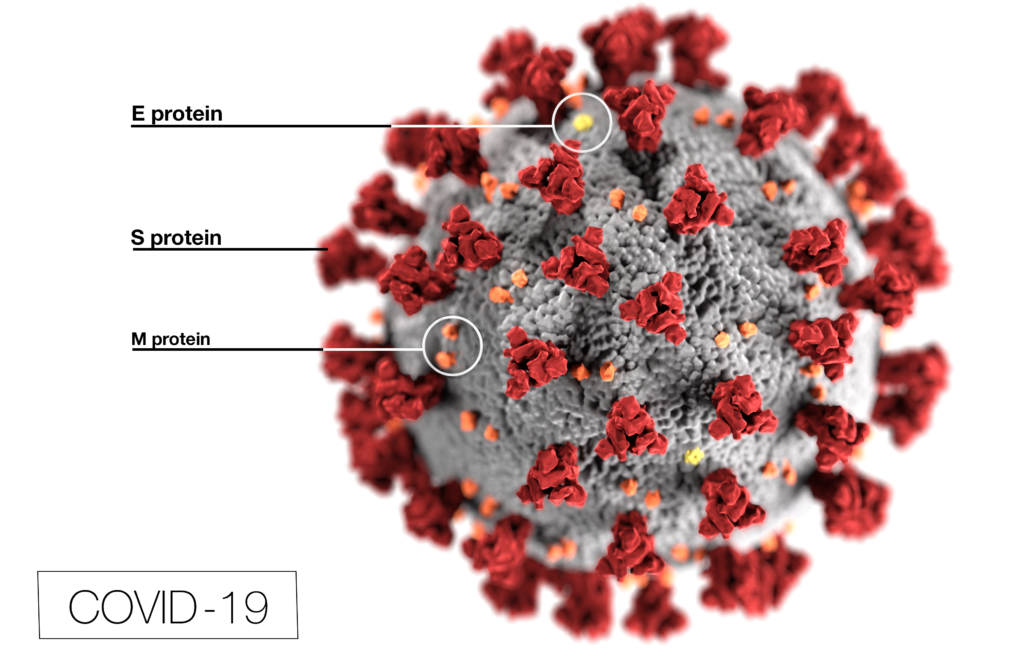
Coronavirus (CoV) researchers are working quickly to understand the entry of SARS-CoV-2 into cells. The Spike or S proteins on the surface of a CoV is trimer. The monomer is composed of an S1 and S2 domain. The division of S1 and S2 happens in the virus producing cell through a furin cleavage site between the two domains. The trimer binds to cell surface proteins. In the case of the SARS-CoV, the receptor is angiotensin converting enzyme 2. (ACE2). The MERS-CoV utilizes the cell-surface dipeptidyl peptidase IV protein. SARS-CoV-2 uses ACE2 as well. Internalized S protein goes though a second cleavage by a host cell protease, near the S1/S2 cleavage site called S2′, which leads to a drastic change in conformation thought to facilitate membrane fusion and entry of the virus into the cell (1).
CDC / Alissa Eckert, MS; Dan Higgins, MAMS
Rather than work directly with the virus, researchers have chosen to make pseudotyped viral particles. Pseudotyped viral particles contain the envelope proteins of a well-known parent virus (e.g., vesicular stomatitis virus) with the native host cell binding protein (e.g., glycoprotein G) exchanged for the host cell binding protein (S protein) of the virus under investigation. The pseudotyped viral particle typically carries a reporter plasmid, most commonly firefly luciferase (FLuc), with the necessary genetic elements to be packaged in the particle.
To create the pseudotyped viral particle, plasmids or RNA alone are transfected into cells and the pseudotyped viruses work their way through the endoplasmic reticulum and golgi to bud from the cells into the culture medium. The pseudoviruses are used to study the process of viral entry via the exchanged protein from the virus of interest. Entry is monitored through assay of the reporter. The reporter could be a luciferase or a fluorescent protein.
Bioluminescent reporter assays are an excellent choice for analyzing gene regulation because they provide higher sensitivity, wider dynamic range and better signal-to-background ratios compared to colorimetric or fluorescent assays. In a typical genetic reporter assay, cells are transfected with a vector that contains the sequence of interest cloned upstream of a reporter gene, and the reporter activity is used to determine how the target sequence influences gene expression under experimental conditions. A second control reporter encoded on the same or a different plasmid is an essential internal control. The secondary reporter is used to normalize the data and compensate for variability caused by differences in cell number, lysis efficiency, cell viability, transfection efficiency, temperature, and measurement time.
Basic Introduction to the Strategy of Reporter Gene Assays
For genetic reporter assays, using a secondary control vector with a weak promoter like PGK or TK to ensures that the control does not interfere with activation of your primary reporter vector. Transfection of high amounts of the control plasmid or putting the control reporter under control of a strong promoter like CMV or SV40 often leads to transcriptional squelching or other interference with the experimental promoter (i.e., trans effects). Reporter assays can also be used to quantitatively evaluate microRNA activity by inserting miRNA target sites downstream or 3´ of the reporter gene. For example, the pmirGLO Dual-Luciferase miRNA Target Expression Vector is based on dual-luciferase technology, with firefly luciferase as the primary reporter to monitor mRNA regulation and Renilla luciferase as a control reporter for normalization.
Here in Technical Services we often talk with researchers who are just starting their project and looking for advice on designing their genetic reporter vector. They have questions like:
How much of the upstream promoter region should be included in the vector?
How many copies of a response element will be needed to provide a good response?
Does the location of the element or surrounding sequence alter gene regulation?
These assays are relatively easy to understand in principle. Use a primary and secondary reporter vector transiently transfected into your favorite mammalian cell line. The primary reporter is commonly used as a marker for a gene, promoter, or response element of interest. The secondary reporter drives a steady level of expression of a different marker. We can use that second marker to normalize the changes in expression of the primary under the assumption that the secondary marker is unaffected by what is being experimentally manipulated.
While there are many advantages to dual-reporter assays, they require careful planning to avoid common pitfalls. Here’s what you can do to avoid repeating some of the common mistakes we see with new users:
Here in Technical Services we often talk with researchers at the beginning of their project about how to carefully design and get started with their experiments. It is exciting when you have selected the Luciferase Reporter Vector(s) that will best suit your needs; you are going to make luminescent cells! But, how do you pick the best way to get the vector into your cells to express the reporter? What transfection reagent/method will work best for your cell type and experiment? Do you want to do transient (short-term) transfections, or are you going to establish a stable cell line?
It’s a question I’m asked probably once a week. “What wavelength do I select on my luminometer when performing a luciferase assay?” The question is a good and not altogether unexpected one, especially for those new to bioluminescent assays. The answer is that in most cases, you don’t and in fact shouldn’t select a wavelength (the exception to this rule is if you’re measuring light emitted in two simultaneous luciferase reactions). To understand why requires a bit of an explanation of absorbance, fluorescence, and luminescence assays, and the differences among them.
Absorbance, fluorescence, and luminescence assays are all means to quantify something of interest, be that a genetic reporter, cell viability, cytotoxicity, apoptosis, or other markers. In principle, they are all similar. For example, a genetic reporter assay is an indicator of gene expression. The promoter of a gene of interest can be cloned upstream of a reporter such as β-galactosidase, GFP, or firefly luciferase. The amount of each of these reporters that is transcribed into mRNA and translated into protein by the cell is indicative of the endogenous expression of the gene of interest.
The stage is set. You’ve spent days setting up this experiment. Your bench is spotless. All the materials you need to finally collect data are laid neatly before you. You fetch your cells from the incubator, add your detection reagents, and carefully slide the assay plate into the luminometer. It whirs and buzzes, and data begin to appear on the computer screen. But wait!
These data are garbage!
Don’t let this dramatic person be you. Here are 8 tips from us on things to watch out for before you start your next luminescent assay. Make sure you’ll be getting good data before wasting precious sample!
I’ve got a set of experiments planned that, if all goes well, will provide me with the answer I have been seeking for months. Plus, my supervisor is eagerly awaiting the results because she needs the data for a grant application, so I don’t want to mess it up. However, I am faced with a choice for my firefly and Renilla luciferase reporter assays: Do I use the Dual-Luciferase® Reporter Assay System or Dual-Glo® Luciferase Assay System? What’s the difference? How do I decide which to use? I’m so confused! Help!
Sound familiar? Not to worry! The choice is not difficult once you know how these assays work and how they differ.
XWe use cookies and similar technologies to make our website work, run analytics, improve our website, and show you personalized content and advertising. Some of these cookies are essential for our website to work. For others, we won’t set them unless you accept them. To learn more about our approach to Privacy we invite you to Read More
By clicking “Accept All”, you consent to the use of ALL the cookies. However you may visit Cookie Settings to provide a controlled consent.
We use cookies and similar technologies to make our website work, run analytics, improve our website, and show you personalized content and advertising. Some of these cookies are essential for our website to work. For others, we won’t set them unless you accept them. To find out more about cookies and how to manage cookies, read our Cookie Policy.
If you are located in the EEA, the United Kingdom, or Switzerland, you can change your settings at any time by clicking Manage Cookie Consent in the footer of our website.
Necessary cookies are absolutely essential for the website to function properly. These cookies ensure basic functionalities and security features of the website, anonymously.
Cookie
Duration
Description
cookielawinfo-checbox-analytics
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Analytics".
cookielawinfo-checbox-functional
11 months
The cookie is set by GDPR cookie consent to record the user consent for the cookies in the category "Functional".
cookielawinfo-checbox-others
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Other.
cookielawinfo-checkbox-advertisement
1 year
The cookie is set by GDPR cookie consent to record the user consent for the cookies in the category "Advertisement".
cookielawinfo-checkbox-necessary
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookies is used to store the user consent for the cookies in the category "Necessary".
cookielawinfo-checkbox-performance
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Performance".
gdpr_status
6 months 2 days
This cookie is set by the provider Media.net. This cookie is used to check the status whether the user has accepted the cookie consent box. It also helps in not showing the cookie consent box upon re-entry to the website.
lang
This cookie is used to store the language preferences of a user to serve up content in that stored language the next time user visit the website.
viewed_cookie_policy
11 months
The cookie is set by the GDPR Cookie Consent plugin and is used to store whether or not user has consented to the use of cookies. It does not store any personal data.
Analytical cookies are used to understand how visitors interact with the website. These cookies help provide information on metrics the number of visitors, bounce rate, traffic source, etc.
Cookie
Duration
Description
SC_ANALYTICS_GLOBAL_COOKIE
10 years
This cookie is associated with Sitecore content and personalization. This cookie is used to identify the repeat visit from a single user. Sitecore will send a persistent session cookie to the web client.
vuid
2 years
This domain of this cookie is owned by Vimeo. This cookie is used by vimeo to collect tracking information. It sets a unique ID to embed videos to the website.
WMF-Last-Access
1 month 18 hours 24 minutes
This cookie is used to calculate unique devices accessing the website.
_ga
2 years
This cookie is installed by Google Analytics. The cookie is used to calculate visitor, session, campaign data and keep track of site usage for the site's analytics report. The cookies store information anonymously and assign a randomly generated number to identify unique visitors.
_gid
1 day
This cookie is installed by Google Analytics. The cookie is used to store information of how visitors use a website and helps in creating an analytics report of how the website is doing. The data collected including the number visitors, the source where they have come from, and the pages visted in an anonymous form.
Advertisement cookies are used to provide visitors with relevant ads and marketing campaigns. These cookies track visitors across websites and collect information to provide customized ads.
Cookie
Duration
Description
IDE
1 year 24 days
Used by Google DoubleClick and stores information about how the user uses the website and any other advertisement before visiting the website. This is used to present users with ads that are relevant to them according to the user profile.
test_cookie
15 minutes
This cookie is set by doubleclick.net. The purpose of the cookie is to determine if the user's browser supports cookies.
VISITOR_INFO1_LIVE
5 months 27 days
This cookie is set by Youtube. Used to track the information of the embedded YouTube videos on a website.
Performance cookies are used to understand and analyze the key performance indexes of the website which helps in delivering a better user experience for the visitors.
Cookie
Duration
Description
YSC
session
This cookies is set by Youtube and is used to track the views of embedded videos.
_gat_UA-62336821-1
1 minute
This is a pattern type cookie set by Google Analytics, where the pattern element on the name contains the unique identity number of the account or website it relates to. It appears to be a variation of the _gat cookie which is used to limit the amount of data recorded by Google on high traffic volume websites.