
I’ve got a set of experiments planned that, if all goes well, will provide me with the answer I have been seeking for months. Plus, my supervisor is eagerly awaiting the results because she needs the data for a grant application, so I don’t want to mess it up. However, I am faced with a choice for my firefly and Renilla luciferase reporter assays: Do I use the Dual-Luciferase® Reporter Assay System or Dual-Glo® Luciferase Assay System? What’s the difference? How do I decide which to use? I’m so confused! Help!
Sound familiar? Not to worry! The choice is not difficult once you know how these assays work and how they differ.
How They Work
The Dual-Luciferase® Reporter Assay System (DLR® Assay) and Dual-Glo® Luciferase Assay System accomplish the same thing: The sequential quantification of two bioluminescent reporters, firefly and Renilla luciferases, from a single sample. Firefly luciferase is a 61kDa monomeric reporter protein that catalyzes the oxidation of beetle luciferin in a reaction that requires ATP, Mg2+ and O2 and produces light (Figure 1). Renilla luciferase is a 36kDa monomeric protein and catalyzes a bioluminescent reaction that uses O2 and coelenterazine. Both are common and convenient reporter genes. In my hypothetical experiments, the firefly luciferase is used as the “experimental” reporter to monitor changes in expression of my gene of interest, while Renilla luciferase is used as the control reporter to normalize results for any interfering factors such as differences in transfection efficiency or cell viability. However, I could have easily designed my experiments the other way around, with Renilla luciferase as the experimental reporter.

Figure 1. Bioluminescent reactions catalyzed by firefly and Renilla luciferases.
The DLR® and Dual-Glo® Assays both include a firefly luciferase reagent (Luciferase Assay Reagent II for the DLR® Assay and Dual-Glo® Luciferase Reagent for the Dual-Glo® Assay) and a Renilla luciferase reagent (Stop & Glo® Reagent for the DLR® Assay and Dual-Glo® Stop & Glo® Reagent for the Dual-Glo® Assay). For both systems, the firefly luciferase reagent is added first, and luminescence is measured before the Stop & Glo® Reagent is added to quench the firefly luciferase signal and initiate the Renilla luciferase reaction. Luminescence is measured a second time to quantify Renilla luciferase activity.
How They Differ
Lysis Buffer
There are important differences between these assays that will determine which is best for my situation. The DLR® Assay includes a separate lysis buffer, the Passive Lysis Buffer, and requires that I prepare cell lysates prior to the assay. A portion of each lysate is used in the DLR® Assay, and the remaining lysate can be stored at –20° for up to 1 month or at –70°C for long-term storage. The Dual-Glo® Assay does not require a separate lysis buffer. Cell lysis components are included in the Dual-Glo® Luciferase Reagent. Simply add an equal volume of Dual-Glo® reagent directly to the cell culture, and after at least 10 minutes to allow cell lysis, measure luminescence.
Signal Half-Life
Another difference is signal half-life. The DLR® Assay generates luminescence that rapidly decreases in intensity. The firefly luciferase signal decreases 50% in approximately 12–15 minutes, and the Renilla luciferase signal decreases 50% in less than 3 minutes. These signal kinetics make measurement of firefly and Renilla luciferase activities difficult if large numbers of samples are measured in 96- or 384-well plates. Lysates must be assayed one at a time in a tube format, or a luminometer equipped with one or two reagent injectors must be used to accommodate the “read-inject-read” format of the DLR® Assay.
In contrast, the Dual-Glo® Assay has stabilized luminescent signals, with firefly and Renilla luciferase signal half-lives of approximately 2 hours. Thus, the Dual-Glo® Assay can be used to measure both firefly and Renilla luminescence in multiwell plates using a plate-reading luminometer, CCD camera or similar imaging device without injectors. (Note: Injectors should not be used with the Dual-Glo® Assay to avoid foaming, which can interfere with luminescence measurements.) Plates containing experimental samples can be batch processed: I can add the Dual-Glo® Luciferase Reagent to all wells of the plate, measure firefly luminescence across the entire plate, add the Dual-Glo® Stop & Glo® Reagent to all wells of the plate, then measure Renilla luminescence across the entire plate. Due to the 2-hour half-life, I don’t have to worry about significant signal loss as wells wait to be read.
Light Output
Finally, the light output will be different. Firefly and Renilla luciferases undergo spontaneous inactivation after generating luminescence. This inactivation causes the “flash”-type kinetics characteristic of the DLR® Assay. To generate a more stable luminescent signal that is amenable to high-throughput measurements, the rate of inactivation and subsequently the rate of catalysis must be slowed. For this reason, the Dual-Glo® Assay results in lower luminescence levels than the DLR® Assay, which was developed for maximal sensitivity. This difference in light output will affect my raw data (i.e., the number of relative light units [RLU]). Luminescence levels measured using the DLR® Assay will be dramatically but proportionately higher (approximately 100-fold) than those using the Dual-Glo® Assay.
The background also will be proportionately higher. However, the final results—the trends, as determined by the increase or decrease in the ratio of luciferase activities—will be the same, so does it really make a difference in the end? Do I really care if my samples yield 100,000RLU or 10,000,000RLU if all of the readings, including the background, are proportionally higher? When I worked as a Promega Technical Service Scientist, I would encounter researchers who were obsessed with high luminometer readings, and my coworkers and I would always joke that if someone wanted higher RLU readings for his raw data, he should just tape a piece of paper with three zeroes next to the display of the luminometer; just make sure that the three zeroes are added to all readings, including the background.
Which Assay Do I Pick?
If you are in the same situation as I am, trying to determine which dual-luciferase assay is right for you, ask yourself the following questions:
How many samples am I processing?
The DLR® Assay requires you to prepare a lysate for each sample, which can be stored for later use. The Dual-Glo® Assay offers the convenience of incorporating the cell lysis step into the first step of the reporter assay and processing samples in a multiwell format but consumes the entire sample.
Do I really need those higher luminometer readings, or can I get by with lower readings if it means I have the convenience of performing my assays in multiwell plates?
The DLR® Assay yields higher luminescence readings, but the shorter signal half-lives makes it less suited to high-throughput measurements than the Dual-Glo® Assay.
What luminometer or other imaging device will I use?
The Dual-Glo® Assay does not require a luminometer with injectors due to the longer signal half-lives (in fact, foaming caused by injectors can interfere with luminescence measurements), while the DLR® Assay requires an imaging device that can measure luminescence within seconds of reagent addition.
Learn more about NanoLuc® Luciferase and its many applications in life-science research.
Now that you have a better understanding of how these two products differ, there’s one more option worth considering: the Nano-Glo® Dual-Luciferase® Reporter Assay System (NanoDLR™). Like the Dual-Glo® Assay, NanoDLR™ has glow signal kinetics. Unlike both the DLR® and Dual-Glo® Assays, the second reporter is NanoLuc® luciferase, an ATP-independent enzyme engineered from a deep-sea shrimp luciferase. NanoLuc is the smallest engineered luciferase available and up to 150x brighter than conventional luciferase. Combined with improved firefly luciferase chemistry, this gives you more sensitive detection of both reporters from a single sample.
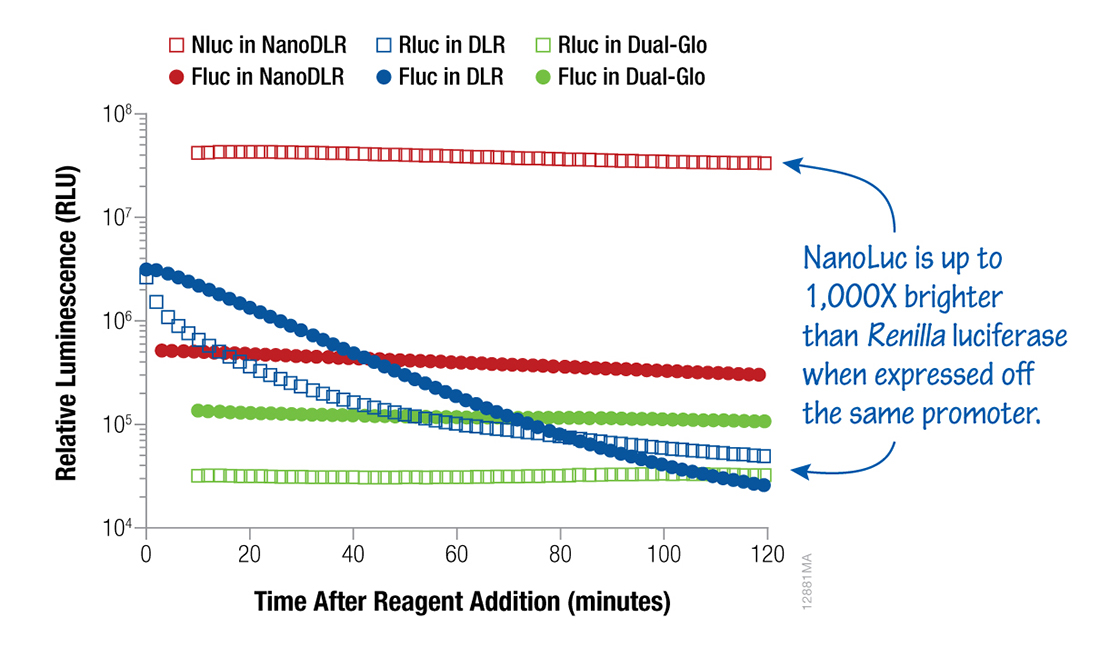
Figure 2. A comparative look at luminescence signals from HEK293 cells transfected with a 1:1:8 ratio of either TK-Rluc (Renilla):TK-Fluc (firefly):carrier DNA or TK-Nluc (NanoLuc):TK-Fluc:carrier DNA and assayed using NanoDLR™, DLR™ or Dual-Glo® Assay Systems, as indicated.
For more information about how these assays work and which one might be best for you, check out the technical literature:
- Dual-Luciferase® Reporter Assay System Technical Manual TM040
- Dual-Glo® Luciferase Assay System Technical Manual TM058
- Nano-Glo® Dual-Luciferase® Reporter Assay Technical Manual TM426
- Dual-Glo™ Luciferase Assay System: Convenient dual reporter measurements in 96- and 384-well plates
im trying to understand more about luciferase assay,someone please guide me to understands this assay,the plasmid that i used are PRLTK and PPRE..i dont understand why we should substract the background to calculate data before making graph.=(
Hello Shadariah,
We have forwarded your question to technical services (techserv@promega.com), and we will post their reply in the blog comments shortly.
Michele
Hi Shadariah,
The background is subtracted the luminescence measurements of both firefly and Renilla luciferases for maximum accuracy of your data analysis. The background luminescence in your experiment should be quite low because neither enzyme is endogenously expressed in mammalian cells, therefore the source of background luminescence is either a characteristic of the luminometer or of the luminescent substrate. Beetle luciferin, one of the firefly luciferase substrates, does not generate light in the absence of luciferase in the Dual-Glo® Reagents. Coelenterate luciferin, the substrate for Renilla luciferase, does autoluminesce but Promega has included a proprietary chemistry in the Dual-Glo® Stop & Glo® Reagent to minimize enzyme-independent luminescence (autoluminsecence). Background measurements for firefly luciferase should be taken from samples consisting of nontransfected cells and Dual-Glo® Luciferase Reagent. For Renilla luciferase, background measurements should be taken from samples containing nontransfected cells and both Dual-Glo® Luciferase Reagent and Dual-Glo® Stop & Glo® Reagent. Sample volumes for background measurements must be the same as experimental sample volumes and contain the same media/sera combinations as the experimental samples. I have attached a Cell Notes Article, titled NORMALIZING GENETIC REPORTER ASSAYS: APPROACHES AND CONSIDERATIONS FOR INCREASING CONSISTENCY AND STATISTICAL SIGNIFICANCE, which you may find to be helpful in explaining the key points to consider for successful data analysis.
Here is a link to an article for more information: http://www.promega.com/resources/articles/pubhub/cellnotes/normalizing-genetic-reporter-assays/
If you have any additional questions, our technical services scientists will be happy to help you at techserv@promega.com
Hi,
How about using Dual-Glo in a 12-well plate, detach the cells with trypsin, wash the cells with PBS, resuspend in 200µl medium/PBS and take an aliquot of 75µl of cells in a 96-well plate and proceed with dual-glo protocol. This way we can have the benefit of preserving an aliquot cells for future use. Please let me know your suggestions..
rsk, I haven’t heard of anyone doing what you propose, but my first reaction is that splitting the cells into aliquots is not ideal. If you don’t need intact cells, I recommend making a lysate, then splitting the lysate. You could simply remove the medium, make a lysate using the Glo Lysis Buffer, use a portion for the Dual Glo Assay and store the remaining lysate at –20°C or –70°C for future use.
If you need aliquots of intact cells, there are a few things to keep in mind with your approach: Splitting the cells will introduce another variable that could affect your raw data (e.g., incomplete trypsinization could result in aliquots with differences in cell number) although you can normalize for any differences using the second reporter. Also, there is a chance that the additional manipulation of the cells could affect either or both reporter activities. The magnitude of this effect might be affected by how fast you split, wash and freeze the cells, the relative response rate of the reporter genes (e.g., are you using wildtype luciferase or one of the rapid response luciferases such as luc2P or luc2CP?) and probably other factors. Most reporters don’t respond quickly enough to allow you to detect changes in reporter activity due to these additional steps, but you might consider a preliminary experiment to show that the extra manipulations do not have an effect.
A very good explanation to beginners in the field, thank you :) wonderfully written
Glad that you found the blog helpful. And, if you have suggestions for more topics that you would like to see us write about, let us know.
Hello Terri,
I am planning to use Luciferase assay to analyze the transcription activation of a gene in Xenopus laevis embryos. We will have to first standarize the dilution of the sample and plasmid injected. I want to know which kit is better for me, the Dual Lucif or the Dual Glo. I like the dual Glo for its stable signal but I want to be able to save sample for future use, and the Dual Luciferase has more Lysis Buffer than the Dual Glo. What should I do. Thank you very much in advance. Please let me know your suggestions..
Dear Santiago Cerrizuela, thank you for your inquiry. I apologize for the delay in our reply. This is Laura at Promega Technical Services. The Glo Lysis Buffer (catalog no. E2661) can be used with the Dual-Glo system if you would like to make lysates first, and then use a portion of the lysate in the Dual-Glo reaction. Regardless if you use the Dual-Glo System or Dual-Luciferase System (which comes with Passive Lysis Buffer), you may have to homogenize the Xenopus embryos in the lysis buffer. The Glo Lysis and Passive Lysis Buffers may not sufficiently lyse the embryos without some mechanical disruption. If you have further questions or need additional information, please contact Promega Technical Services at techserv@promega.com.
I tried luciferase assay in mice adipose tissue,but i did,t saw any cellular pellet after cell lysis with lysis buffer and shaken for 20 minutes. Although i rubbed the cell plate for manually detaching the cells and then performed the assay but i did,t got any good result of luciferase assay. Need your guidance please
Dear Rajwali Khan, thank you for your inquiry. My name is Mailee from Promega Technical Services. Please email me at TechServ@promega.com with additional information on your experiment. I would be happy to help you troubleshoot your luciferase assay using mice adipose tissue.
Hello..I am using Nano-Glo Dual-Luciferase Reporter Assay System (REF:N1620)
I wonder how many maximum time freeze-thaw the kit is recommended?
Hello,You can store the entire kit at –20°C. The lyophilized substrate and the NanoDLR Stop&Glo substrate would be kept at -20C until use. Prior to reconstitution, the two buffers are very stable, especially the ONE-Glo EX buffer. We say that the ONE-Glo EX buffer can be kept at 4°C 1 year or RT for 6 months. For the NanoDLR Stop&Glo buffer, is also very stable and can be kept at 4°C for 6 month or room temperature for 3 months. There is really no reason to freeze/thaw the buffers repeatedly because you can leave them at 4°C once thawed.For reconstituted reagents with the substrates added, NanoDLR was designed to have extended stability compared to similar reagents. At 4C, the reconstituted ONE-Glo EX should maintain 50% of its original activity after 1 month. The best thing to do is to dispense into smaller, single-use aliquots, rather than continuing the thaw and re-freeze the entire product. We recommend making the reconstituted NanoDLR Stop&Glo reagent fresh, for each experiment in the amount needed.
Hello,
can the Nano-Glo® Dual-Luciferase® Reporter Assay (N1630) be used with Renilla luciferase instead of NanoLuc® as constitutive control as well?
Thank you in advance!
Hi there, I have forwarded your question to technical services and will post their reply here. Michele
Unfortunately, Renilla luciferase (Rluc) will not work with Nano-Glo® Dual-Luciferase® Reporter Assay. This all comes down to substrate specificity. Rluc uses coelenterazine as a substrate, but NanoLuc® luciferase is a furimazine substrate. Because of this, a firefly/Rluc system should be measured using the Dual-Glo® Luciferase Assay System (E2920) or Dual-Luciferase® Reporter Assay System (E1910).
In a pinch, all three of the dual kits can be used to measure firefly luminescence, but the second reporter must match the kit.
Please let us know if you have any additional questions.
Hi,
I am using Steady-Glo® Luciferase Assay System (Reference Number: E2510) for evaluating luciferase trasnfection from mesenchymal stem cells. I have two questions, and I’d feel so pleasent if you could help me. First, I would like to know if I could aliquotate and store the reconstituted Steady-Glo® Assay Reagent at -80°C, and for up time? (The manual usser only does a mention of storing it at -20 for up 2 weeks). On the other hand, I need to measure the total protein within the lysate, and due to the lysis buffer and substrate is in a single reagent, I would like to know if it is possible to store the remaining extract, which already incluide the Steady-Glo® Assay Reagent, at -70°C for doing a bradford or any other assay for measuting total proteins later. Would the presence of Steady-Glo® Assay Reagent affect the total protein measure?
Thank yoy for the help and the advises.
Regards,
Leslie
Dear Leslie,
My name is Monica and I am part of the Promega Technical Services team. It is my pleasure to answer your blog post inquiry about Steady-Glo® assay! I will try to address each of your questions in detail below.
Steady-Glo® Reagent does not have very good long-term storage stability; once prepared it will slowly lose activity over time. If stored at -80°C we recommend using Steady-Glo® Reagent within 3-4 weeks after it has been reconstituted. It is possible to aliquot the reconstituted Steady-Glo® Reagent, though this may not be necessary depending on the volume of reagent you prepared and how many assay plates you need to run. There is always a risk when aliquoting that an unintentional gradient is formed if the reagent is not mixed well before (and even during) the aliquoting process. Keep in mind that Steady-Glo® Reagent can withstand up to five freeze/thaw cycles without significant loss in activity. Knowing this, you can decide whether or not it is worthwhile to aliquot the reconstituted Steady-Glo® Reagent or plan to re-freeze after each use.
It is possible to lyse your cells with the Steady-Glo® Reagent and then use this lysate in a protein detection assay downstream (e.g., Bradford assay). We have tested this before and found that for Steady-Glo® it is best to dilute the lysate 0.001X beforehand, and use a protein assay that is specifically compatible with detergents and reducing agents. This article on our website discusses this in greater detail and provides some protein assay kit suggestions:
“Compatibility of the Pierce BCA Protein Assay with Promega Lysis Buffers and Lytic Assay Reagents”
https://www.promega.com/resources/pubhub/enotes/compatibility-of-pierce-bca-protein-assay-with-promega-lysis-buffers-and-lytic-reagents/
As a side note, when performing the protein detection assay you will want to make sure you prepare standards and blanks in the same way as your test samples (i.e., in diluted Steady-Glo® Reagent) so that you are accounting for the background signal appropriately.
I do not believe we have tested the protein stability of cell lysates from after performing Steady-Glo® assay, so I am afraid I cannot provide you with concrete recommendations here. Based on my experience and discussions with other colleagues, it seems reasonable to store the cell lysate at -80°C and perform the protein detection assay later without risk of too much protein degradation. Just make sure to minimize the freeze/thaw cycles of the lysate, and do not plan to store this lysate for too long (e.g., <1 month) since no stabilizers have been added.
I hope that this information is helpful. Please let me know if you have any other questions or concerns that I can assist you with. Good luck with your work and have a nice day!
Best regards,
Monica
Hi,
I am using Steady-Glo® Luciferase Assay System (Reference Number: E2510) for evaluating luciferase trasnfection from mesenchymal stem cells. I have two questions, and I’d feel so pleasent if you could help me. First, I would like to know if I could aliquotate and store the reconstituted Steady-Glo® Assay Reagent at -80°C, and for how long? (The manual usser only does a mention of storing it at -20 for up 2 weeks). On the other hand, I need to measure the total protein within the lysate, and due to the lysis buffer and substrate is in a single reagent, I would like to know if it is possible to store the remaining extract, which already incluide the Steady-Glo® Assay Reagent, at -70°C for doing a bradford or any other assay for measuring total proteins later. Would the presence of Steady-Glo® Assay Reagent affect the total protein measure?
Thank yoy for the help and the advises.
Regards,
Leslie
Hi,
I’m planning to use Dual Glo kit for my luciferase assay. I would like to know whether I can culture cell in a 6 cm dish instead of directly culture cell in 96 well plate, then trypsinize the cell and add equal volume of cell into 96 -well plate for luminescence assay as stated in protocol. Or if I need to culture cell in a 6 cm dish and perform the assay in 96-well plate, how should I lyse the cell (which kit/reagent should be used)? Thanks.
Best regards,
Elly
It is okay to culture the cells in a 6-cm dish, trypsinize and collect the cells, and then replate the cells into a 96-well plate to set up the reporter assay. However, we would not recommend this approach if the cells are cultured and treated in the 6-cm dish (i.e., only transferring the cells into a 96-well plate to measure the luciferase activities), as the process of trypsinization and replating can lead to increased variability in assay performance. If it is more preferable to culture and treat the cells in the 6-cm dish before measuring the luciferase activities, then we would suggest creating a cell lysate using the Glo Lysis Buffer (Cat.# E2661). The lysate can be assayed with the Dual-Glo® assay according to the Technical Manual for the assay.
Please note that the Glo Lysis Buffer is not optimized for the Dual-Glo® Luciferase Assay System and the use of Glo Lysis Buffer can cause a decrease a signal half-life for the Dual-Glo® assay. Having said that, the signal half-life for both luciferases should still be at least one hour when Glo Lysis Buffer is used with the Dual-Glo® assay.
Lastly, I would like to note that, when following our recommendation for the volume of Glo Lysis Buffer to use for various plate sizes, the lysate created will be more concentrated. Please be mindful of not saturating the Dual-Glo® assay or the luminometer when assaying the lysate.
If you have further questions, our technical services scientists are available to help at https://www.promega.com/support/