Could bacteria survive on Mars? While images of the red planet might spark thoughts of barren landscapes and lifeless deserts, Mars holds a fascinating possibility: under suitable conditions, pockets of salty, perchlorate-rich brines could temporarily form on or near its surface. These brines are formed by salts that naturally absorb water from their surroundings. By lowering the temperature at which water freezes, these salts can stabilize liquid water, raising intriguing questions about the potential for microbial life. But what exactly would it take for bacteria to survive there? New research from Kloss et al. published in Scientific Reports sheds light on this cosmic question.
On November 15, 2021, Science Advances announced the launch of The Human Proteoform Project. The ambitious project, led by the Consortium for Top-Down Proteomics, aims to address a critical next step in disease research. This means developing new technologies to outline a complete set of protein forms based on the ~20,000 genes in the human genome.
Mass spectrometry depends on the successful digestion of proteins using proteases. Many commercially available proteomic-grade trypsins contain natural contaminants that produce non-specific cleavages. Trypsin Platinum, a new protease from Promega provides maximum specificity, giving you cleaner and more conclusive data from mass spec.
Trypsin is typically extracted from bovine or porcine pancreas. In addition to trypsin, both of these sources also contain chymotrypsin. To suppress chymotryptic activity, trypsin is treated with tosyl phenylalanyl chloromethyl ketone, or TPCK, to irreversibly inhibit the chymotrypsin. However, trace amounts of chymotrypsin appear to escape this inhibition and produce non-specific cleavages, as seen in the figure below.
With the use of a suite of “-omics” technologies you can examine the way in which complex cellular processes work together across all molecular domains (i.e., proteomics, metabolomics, transcriptomics) in a single biological system. Several studies have been published across a wide range of fields illustrating the power of such a unified approach (1,2). Most studies however did not focus on the development of a high-throughput, unified sample preparation approach to complement high-throughput “omic” analytics.
A recent publication by Gutierrez and colleagues presents a simple high-throughput process (SPOT) that has been optimized to provide high-quality specimens for metabolomics, proteomics, and transcriptomics from a common cell culture sample (3). They demonstrate that this approach can process 16−24 samples from a cell pellet to a desalted sample ready for mass spectrometry analysis within 9 hours. They also demonstrated that the combined process did not sacrifice the quality of data when compared to individual sample preparation methods.
Bottom-up proteomics focuses on the analysis of protein mixtures after enzymatic digestion of the proteins into peptides. The resulting complex mixture of peptides is analyzed by reverse-phase liquid chromatography (RP-LC) coupled to tandem mass spectrometry (MS/MS). Identification of peptides and subsequently proteins is completed by matching peptide fragment ion spectra to theoretical spectra generated from protein databases.
Trypsin has become the gold standard for protein digestion to peptides for shotgun proteomics. Trypsin is a serine protease. It cleaves proteins into peptides with an average size of 700-1500 daltons, which is in the ideal range for MS (1). It is highly specific, cutting at the carboxyl side of arginine and lysine residues. The C-terminal arginine and lysine peptides are charged, making them detectable by MS. Trypsin is highly active and tolerant of many additives.
Even with these technical features, the use of trypsin in bottom-up proteomics may impose certain limits in the ability to grasp the full proteome, Tightly-folded proteins can resist trypsin digestion. Post-translational modifications (PTMs) present a different challenge for trypsin because glycans often limit trypsin access to cleavage sites, and acetylation makes lysine and arginine residues resistant to trypsin digestion.
Are you looking for proteases to use in your research? Explore our portfolio of proteases today.
To overcome these problems, the proteomics community has begun to explore alternative proteases to complement trypsin. However, protocols, as well as expected results generated when using these alternative proteases have not been systematically documented.
In a recent reference (2), optimized protocols for six alternative proteases that have already shown promise in their applicability in proteomics, namely chymotrypsin, Lys-C, Lys-N, Asp-N, Glu-C and Arg-C have been created.
Data describe the appropriate MS data analysis methods and the anticipated results in the case of the analysis of a single protein (BSA) and a more complex cellular lysate (Escherichia coli). The digestion protocol presented here is convenient and robust and can be completed in approximately in 2 days.
Try a sample of high-efficiency Trypsin Platinum today!
Visit our website for more on Trypsin Platinum, Mass Spectrometry Grade, with enhanced proteolytic efficiency and superior autoproteolytic resistance.
Artist’s conception of Mimivirus structure, the first of the giant viruses identified.
Following the discovery of Mimivirus (1) the first virus with a particles large enough to be visible under the light microscope, two additional “giant” viruses infecting Acanthamoeba have been discovered Pandoravirus (2) and Pithovirus sibericum (3), the latter from a 30,000 year old Siberian permafrost. A fourth type was recently isolated from the same sample of permafrost by Legendre et al, and named Mollivirus sibericum (4).
Mollivirus sibericum has an approximately spherical virion (0.6 µm diameter) with a 651kb GC-rich genome that encodes 523 proteins. To further characterize the virus the researchers performed transcromic- and proteomic-based time course experiments.
For the particle proteome and infectious cycle analysis, proteins were extracted and then run a 4–12% polyacrylamide gel, and trypsin digests were performed in-gel before nano LC-MS/MS analysis of the resulting peptides. Proteomic studies of the particle showed that it lacked an embarked transcription apparatus, but revealed an unusual presence of many ribosomal and ribosome-related proteins.
When the researchers explored the proteome during the course of an entire infectious cycle, the relative proportions of Mollivirus-, mitochondrion-, and Acanthamoeba encoded proteins were found to vary consistently with an infectious pattern that preserved the cellular host integrity as long as possible and with the release of newly formed virus particles through exocytosis.
In an interesting footnote, the authors of this study point out the fact that two different viruses retain their infectivity in prehistorical permafrost layers should be a concern in the context of global warming and the potential to expose humans to primeval viruses.
Filter-aided sample preparation (FASP) method is used for the on-filter digestion of proteins prior to mass-spectrometry-based analyses (1,2). FASP was designed for the removal of detergents, and chaotropes that were used for sample preparation. In addition, FASP removes components such as salts, nucleic acids and lipids. Akylation of reduced cysteine residues is also carried out on filter, after which protein is proteolyzed by use of trypsin on filter in the optimal buffer of the enzyme. Subsequent elution and desalting of the peptide-rich solution then provides a sample ready for LC–MS/MS analysis.
Erde et al. (3) described an enhanced FASP (eFASP) workflow that included 0.2% DCA in the exchange, alkylation, and digestion buffers,thus enhancing trypsin proteolysis, resulting in increases cytosolic and membrane protein representation. DCA has been reported (4) to improve the efficiency of the denaturation, solubilization, and tryptic digestion of proteins, particularly proteolytically resistant myoglobin and integral membrane proteins, thereby enhancing the efficiency of their identification with regard to the number of identified proteins and unique peptides.
In a recent publication (5) traditional FASP and eFASP were re-evaluated by ultra-high-performance liquid chromatography coupled to a quadrupole mass filter Orbitrap analyzer (Q Exactive). The results indicate that at the protein level, both methods extracted essentially the same number of hydrophobic transmembrane containing proteins as well as proteins associated with the cytoplasm or the cytoplasmic and outer membranes.
The LC–MS/MS results indicate that FASP and eFASP showed no significant differences at the protein level. However, because of the slight differences in selectivity at the physicochemical level of peptides, these methods can be seen to be somewhat complementary for analyses of complex peptide mixtures.
N-Glycosylation is a common protein post-translational modification occurring on asparagine residues of the consensus sequence asparagine-X-serine/threonine, where X may be any amino acid except proline. Protein N-glycosylation takes place in the endoplasmic reticulum (ER) as well as in the Golgi apparatus.
Approximately half of all proteins typically expressed in a cell undergo this modification, which entails the covalent addition of sugar moieties to specific amino acids. There are many potential functions of glycosylation. For instance, physical properties include: folding, trafficking, packing, stabilization and protease protection. N-glycans present at the cell surface are directly involved in cell−cell or cell−protein interactions that trigger various biological responses.
The standard method used to profile the N-glycosylation pattern of cells is glycoprotein isolation followed by denaturation and/or tryptic digestion of the glycoproteins and an enzymatic release of the N-glycans using PNGase F followed by analysis mass spec. This method has been reported to yield high levels of high-mannose N-glycans that stem from both membrane proteins as well as proteins from the ER.(1,2)
Are you looking for proteases to use in your research? Explore our portfolio of proteases today.
For those researchers interested in characterizing only cell surface glycans (i.e., complex N-glycans) a recent reference has developed a model system using HEK-292 cells that demonstrates a reproducible, sensitive, and fast method to profile surface N-glycosylation from living cells (3). The method involves standard centrifugation followed by enzymatic release of cell surface N-glycans. When compared to the standard methods the detection and quantification of complex-type N-glycans by increased their relative amount from 14 to 85%.
Proteomics, the analysis of the entire protein content of a living system, has become a vital part of life science research, and mass spectrometry (MS) is the method for analyzing proteins. MS analysis of protein content allows researchers to identify proteins, sequence them and determine the nature of post translational modifications.
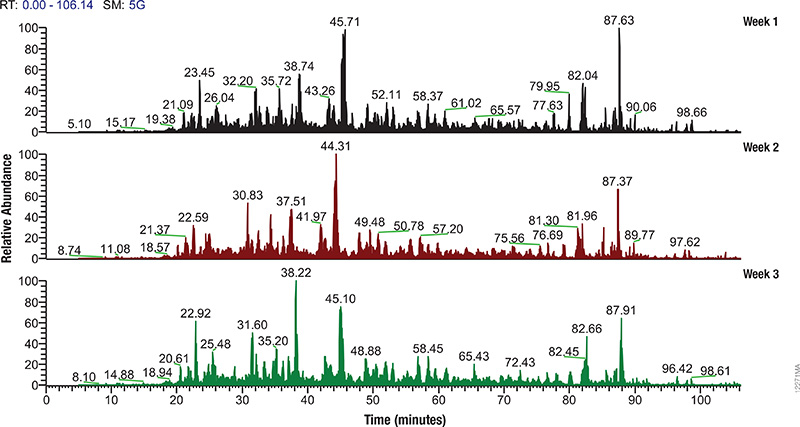
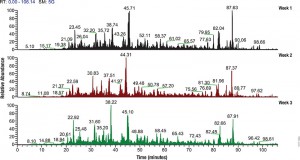
LC/MS performance monitoring. Each run used 1μg of human predigested protein extract injected into the instrument (Waters NanoAquity HPLC System interfaced to a Thermo Fisher Q Exactive™ Hybrid Quadrupole-Orbitrap Mass Spectrometer). Peptides were resolved with a 2-hour gradient. Weekly monitoring with the human extract ensured consistent analytical performance of the instrument.
Mass spectrometry allows characterization of molecules by converting them to ions so that they can be manipulated in electrical and magnetic fields. Basically a small sample (analyte) is ionized, usually to cations by loss of an electron. After ionization, the charged particles (ions) are separated by mass and charge; the separated particles are measured and data displayed as a mass spectrum. The mass spectrum is typically presented as a bar graph where each peak represents a single charged particle having a specific mass-to-charge (m/z) ratio. The height of the peak represents the relative abundance of the particle. The number and relative abundance of the ions reveal how different parts of the molecule relate to each other.
For the study of large, organic macromolecules, matrix associated laser desorption/ionization (MALDI) or tandem mass spec/collision induced dissociation (MS/MS) techniques are often used to generate the charged particles from the analyte. MS analysis brings sensitivity and specificity to proteome analysis. The technique has excellent resolution and is able to distinguish one ion from another, even when their m/z ratios are similar. Macromolecules are present in extremely different concentrations in the cells, and MS analysis can detect biomolecules across five logs of concentration.
The complexity of biological samples places high demand on mass spec analytical capability. Adequate monitoring of instrument performance for proteomics studies requires equally complex reference material such as whole-cell extracts. However, whole-cell extracts available commercially are developed for general research (e.g., enzymatic or Western blot analysis) and contain detergents and salts that interfere with reverse phase liquid chromatography and mass spectrometry. Even after clean up, the extracts need to be digested, requiring time, labor and experience to generate samples for use in mass spectrometry. To address the need for complex protein material, we have developed whole-cell protein extracts from yeast and human cells. The yeast extract offers the convenience of a relatively small and well annotated proteome, whereas the human extract provides a complex proteome with large dynamic range. The human extract also serves as reference material for studies targeting the human proteome.
The extracts are free of compounds that interfere with reverse phase liquid chromatography-mass spectrometry (LC-MS), and have been reduced with DTT and alkylated with iodoacetamide then digested with Trypsin/Lys-C Mix and lyophilized. These digested extracts (tryptic peptides) can be readily reconstituted in trifluoroacetic acid (TFA) or formic acid and injected into an instrument. The same human and yeast whole-cell extracts also are provided in an intact (undigested) form for users who would like to develop an independent method for preparing protein mass spectrometry samples. For convenience, the intact extracts are provided as a frozen solution.
Consistent extract protein composition is ensured by tight control over cell culture conditions and manufacturing process. Lot-to-lot consistency of extracts is monitored by various protein and peptide qualitative and quantitation methods, including LC-MS. (Quality control results are provided upon request.) Our manufacturing process assures compatibility with reverse phase liquid chromatography and mass spectrometry, minimal nonspecific protein fragmentation, nonbiological post-translational modifi cations and,for digested extracts, minimal undigested peptides. The extracts are optimized for a high number of peptide and protein identifications in mass spectrometry analysis.
XWe use cookies and similar technologies to make our website work, run analytics, improve our website, and show you personalized content and advertising. Some of these cookies are essential for our website to work. For others, we won’t set them unless you accept them. To learn more about our approach to Privacy we invite you to Read More
By clicking “Accept All”, you consent to the use of ALL the cookies. However you may visit Cookie Settings to provide a controlled consent.
We use cookies and similar technologies to make our website work, run analytics, improve our website, and show you personalized content and advertising. Some of these cookies are essential for our website to work. For others, we won’t set them unless you accept them. To find out more about cookies and how to manage cookies, read our Cookie Policy.
If you are located in the EEA, the United Kingdom, or Switzerland, you can change your settings at any time by clicking Manage Cookie Consent in the footer of our website.
Necessary cookies are absolutely essential for the website to function properly. These cookies ensure basic functionalities and security features of the website, anonymously.
Cookie
Duration
Description
cookielawinfo-checbox-analytics
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Analytics".
cookielawinfo-checbox-functional
11 months
The cookie is set by GDPR cookie consent to record the user consent for the cookies in the category "Functional".
cookielawinfo-checbox-others
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Other.
cookielawinfo-checkbox-advertisement
1 year
The cookie is set by GDPR cookie consent to record the user consent for the cookies in the category "Advertisement".
cookielawinfo-checkbox-necessary
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookies is used to store the user consent for the cookies in the category "Necessary".
cookielawinfo-checkbox-performance
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Performance".
gdpr_status
6 months 2 days
This cookie is set by the provider Media.net. This cookie is used to check the status whether the user has accepted the cookie consent box. It also helps in not showing the cookie consent box upon re-entry to the website.
lang
This cookie is used to store the language preferences of a user to serve up content in that stored language the next time user visit the website.
viewed_cookie_policy
11 months
The cookie is set by the GDPR Cookie Consent plugin and is used to store whether or not user has consented to the use of cookies. It does not store any personal data.
Analytical cookies are used to understand how visitors interact with the website. These cookies help provide information on metrics the number of visitors, bounce rate, traffic source, etc.
Cookie
Duration
Description
SC_ANALYTICS_GLOBAL_COOKIE
10 years
This cookie is associated with Sitecore content and personalization. This cookie is used to identify the repeat visit from a single user. Sitecore will send a persistent session cookie to the web client.
vuid
2 years
This domain of this cookie is owned by Vimeo. This cookie is used by vimeo to collect tracking information. It sets a unique ID to embed videos to the website.
WMF-Last-Access
1 month 18 hours 24 minutes
This cookie is used to calculate unique devices accessing the website.
_ga
2 years
This cookie is installed by Google Analytics. The cookie is used to calculate visitor, session, campaign data and keep track of site usage for the site's analytics report. The cookies store information anonymously and assign a randomly generated number to identify unique visitors.
_gid
1 day
This cookie is installed by Google Analytics. The cookie is used to store information of how visitors use a website and helps in creating an analytics report of how the website is doing. The data collected including the number visitors, the source where they have come from, and the pages visted in an anonymous form.
Advertisement cookies are used to provide visitors with relevant ads and marketing campaigns. These cookies track visitors across websites and collect information to provide customized ads.
Cookie
Duration
Description
IDE
1 year 24 days
Used by Google DoubleClick and stores information about how the user uses the website and any other advertisement before visiting the website. This is used to present users with ads that are relevant to them according to the user profile.
test_cookie
15 minutes
This cookie is set by doubleclick.net. The purpose of the cookie is to determine if the user's browser supports cookies.
VISITOR_INFO1_LIVE
5 months 27 days
This cookie is set by Youtube. Used to track the information of the embedded YouTube videos on a website.
Performance cookies are used to understand and analyze the key performance indexes of the website which helps in delivering a better user experience for the visitors.
Cookie
Duration
Description
YSC
session
This cookies is set by Youtube and is used to track the views of embedded videos.
_gat_UA-62336821-1
1 minute
This is a pattern type cookie set by Google Analytics, where the pattern element on the name contains the unique identity number of the account or website it relates to. It appears to be a variation of the _gat cookie which is used to limit the amount of data recorded by Google on high traffic volume websites.




 Filter-aided sample preparation (FASP) method is used for the on-filter digestion of proteins prior to mass-spectrometry-based analyses (1,2). FASP was designed for the removal of detergents, and chaotropes that were used for sample preparation. In addition, FASP removes components such as salts, nucleic acids and lipids. Akylation of reduced cysteine residues is also carried out on filter, after which protein is proteolyzed by use of trypsin on filter in the optimal buffer of the enzyme. Subsequent elution and desalting of the peptide-rich solution then provides a sample ready for LC–MS/MS analysis.
Filter-aided sample preparation (FASP) method is used for the on-filter digestion of proteins prior to mass-spectrometry-based analyses (1,2). FASP was designed for the removal of detergents, and chaotropes that were used for sample preparation. In addition, FASP removes components such as salts, nucleic acids and lipids. Akylation of reduced cysteine residues is also carried out on filter, after which protein is proteolyzed by use of trypsin on filter in the optimal buffer of the enzyme. Subsequent elution and desalting of the peptide-rich solution then provides a sample ready for LC–MS/MS analysis.