Livestock, sustainable and herd of cattle on a farm in the countryside for eco friendly environment. Agriculture, animals and cows for meat, dairy or beef trade production industry in a grass field
Veterinary medicine increasingly demands the same analytical rigor applied in human clinical research. Whether researchers are characterizing the protein composition of an equine regenerative therapy, detecting drug residues in livestock or profiling pathogen virulence factors in production animals, the questions are complex, and the tools must be equal to them.
Post-translational modifications of proteins are critical for proper protein function. Modifications such as phosphorylation/dephosphorylation can act as switches that activate or inactivate proteins in signaling cascades. The addition of specific sugars to membrane proteins on cells are critical for recognition, interaction with the extracellular matrix and other activities. While we know volumes about some types of protein modifications, ADP-ribosylation on aspartate and glutamate residues has been more difficult to study because of the chemical instability of these ester-linked modifications.
Matić Lab (Eduardo José Longarini and Ivan Matić) recently published a study that explored mono-ADP-ribosylation (ADPr) on aspartate and glutamate residues by the protein PARP1 and its potential reversal by PARG. PARP1 and PARG signaling are central to DNA repair and apoptosis pathways, making them potentially powerful therapeutic targets in cancer or neurodegenerative diseases in which DNA repair processes are often disrupted.
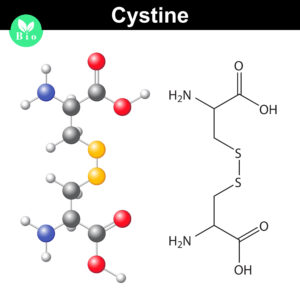
Biologic therapeutics such as monoclonal antibodies and biosimilars are complex proteins that are susceptible to post-translational modifications (PTMs). These chemical modifications can affect the performance and activity of the biologic, potentially resulting in decreased potency and increased immunogenicity. Such modifications include glycosylation, deamidation, oxidation and disulfide bond shuffling. These PTMs can be signs of protein degradation, manufacturing issues or improper storage. Several of these modifications are well characterized, and methods exist for detecting them during biologic manufacture. However, disulfide shuffling is not particularly well characterized for biologics, and no methods exist to easily detect and quantify disulfide bond shuffling in biologics.
Disulfide bonds are important for protein conformation and function
Normally the cysteines in a protein will pair with a predictable or “normal” partner residue either within a polypeptide chain or between two polypeptide chains when they form disulfide bonds. These normal disulfide bonds are important for final protein conformation and stability. Indeed, disulfide bonds are considered an important quality indicator for biologics.
In a recently published study, Coghlan and colleagues designed a semi-automated method for characterizing disulfide bond shuffling on two IgG1 biologics: rituximab (originator drug Rituxan® and biosimilar Acellbia®) and bevacizumab (originator Avastin® and biosimilar Avegra®).
Can pre-digestion with trypsin improve mass spec analysis?
The trypsin protease cleaves proteins on the carboxyterminus of Arginine (Arg) and Lysine (Lys). This cleavage reaction leaves a positive charge on the C-terminus of the resulting peptide, which enhances mass spectrometry analysis (1,2). Because of this advantage, trypsin has become the most commonly used protease for mass spectrometry analysis. Other proteases which cleave differently from trypsin, yielding complementary data are also used in mass spec analysis: these include Asp-N and Glu-C , which cleave acidic residues, and chymotrypsin which cleaves at aromatic residues. The broad spectrum protease, proteinase K is also used for some proteomic analyses. In a recent study, Dau and colleagues investigated whether sequential digestion with trypsin followed by the complementary proteases could improve protein digests for mass spectrometry analysis.
Mass spectrometry depends on the successful digestion of proteins using proteases. Many commercially available proteomic-grade trypsins contain natural contaminants that produce non-specific cleavages. Trypsin Platinum, a new protease from Promega provides maximum specificity, giving you cleaner and more conclusive data from mass spec.
Trypsin is typically extracted from bovine or porcine pancreas. In addition to trypsin, both of these sources also contain chymotrypsin. To suppress chymotryptic activity, trypsin is treated with tosyl phenylalanyl chloromethyl ketone, or TPCK, to irreversibly inhibit the chymotrypsin. However, trace amounts of chymotrypsin appear to escape this inhibition and produce non-specific cleavages, as seen in the figure below.
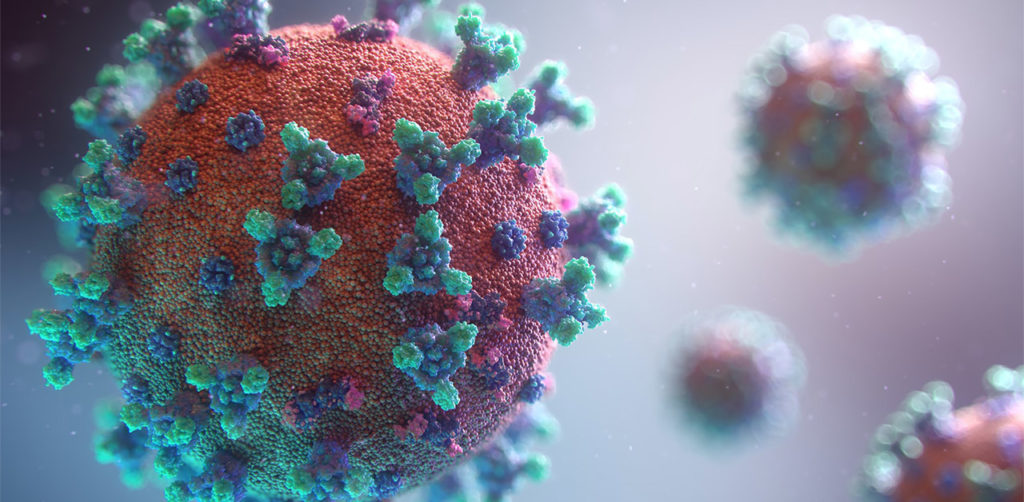
The spike protein of the SARS-CoV-2 virus is a very commonly researched target in COVID-19 vaccine and therapeutic studies because it is an integral part of host cell entry through interactions between the S1 subunit of the spike protein with the ACE2 protein on the target cell surface. Viral proteins important in host cell entry are typically highly glycosylated. Looking at the sequence of the SARS-CoV-2 virus, researchers predict that the spike protein is highly glycosylated. In a recent study, researchers conducted a glycosylation analysis of SARS-CoV-2 proteins using mass spec analysis to determine the N-glycosylation profile of the subunits that make up the spike protein.
Glycans assist in protein folding and help the virus avoid immune recognition by the host. Glycosylation can also have an impact on the antigenicity of the virus, as well as potential effects on vaccine safety and efficacy. Mass spectrometry is widely used for viral characterization studies of influenza viruses. Specifically, mass spec has been used to study influenza protein glycosylation, antigen quantification, and determination of vaccine potency.
In older people, low muscle mass is strongly associated with reduced functional capacity and an increased risk of disability. Myostatin is a negative regulator of muscle growth and has become an important target for pharmaceutical companies designing therapeutics to address age-associated muscle loss.
Anti-myostatin drugs increase muscle size and strength in preclinical studies. Fortetropin is a proteo-lipid complex made from fertilized egg yolk and shows anti-myostatin activity. When Fortetropin is provided as a supplement, lowered circulating myostatin levels are observed both in rodents and in young men. Fortetropin in combination with resistance exercise also lowers myostatin and increased lean body mass.
Sometimes, when using trypsin to study a protein sequence or protein modifications, sequence coverage just isn’t quite as complete as you’d like. Looking for a protease with novel cleavage specificity or a protease that functions well in a low pH environment? Promega has a protease for that.
ProAlanase is a new site-specific endoprotease that preferentially cleaves proteins on the C-terminal side of proline and alanine amino acids. The unique cleavage specificity of ProAlanase (also known as An-PEP or EndoPro; 1–3) can help to uncover parts of the proteome not previously accessible with proteases typically used in proteomic studies.
Glycosylation is the process by which a carbohydrate is covalently attached to target macromolecules, typically proteins. This modification serves various functions including guiding protein folding (1,2), promoting protein stability (2), and participating signaling functions (3).
Ribbon Structure of SARS-CoV-2 Spike Protein
SARS-CoV-2 utilizes an extensively glycosylated spike (S) protein that protrudes from the viral surface to bind to angiotensin-converting enzyme 2 (ACE2) to mediate host-cell entry. Vaccine development has been focused on this protein, which is the focus of the humoral immune response. Understanding the glycan structure of the SARS-CoV-2 virus spike (S) protein will be critical in the development of glycoprotine-based vaccine candidates.
The use of mass spectrometry for the characterization of individual or complex protein samples continues to be one of the fastest growing fields in the life science market.
Bottom-up proteomics is the traditional approach to address these questions. Optimization of each the individual steps (e.g. sample prep, digestion and instrument performance) is critical to the overall success of the entire experiment.
To address issues that may arise in your experimental design, Promega has developed unique tools and complementary webinars to help you along the way.
Here you can find a summary of individual webinars for the following topics:
XWe use cookies and similar technologies to make our website work, run analytics, improve our website, and show you personalized content and advertising. Some of these cookies are essential for our website to work. For others, we won’t set them unless you accept them. To learn more about our approach to Privacy we invite you to Read More
By clicking “Accept All”, you consent to the use of ALL the cookies. However you may visit Cookie Settings to provide a controlled consent.
We use cookies and similar technologies to make our website work, run analytics, improve our website, and show you personalized content and advertising. Some of these cookies are essential for our website to work. For others, we won’t set them unless you accept them. To find out more about cookies and how to manage cookies, read our Cookie Policy.
If you are located in the EEA, the United Kingdom, or Switzerland, you can change your settings at any time by clicking Manage Cookie Consent in the footer of our website.
Necessary cookies are absolutely essential for the website to function properly. These cookies ensure basic functionalities and security features of the website, anonymously.
Cookie
Duration
Description
cookielawinfo-checbox-analytics
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Analytics".
cookielawinfo-checbox-functional
11 months
The cookie is set by GDPR cookie consent to record the user consent for the cookies in the category "Functional".
cookielawinfo-checbox-others
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Other.
cookielawinfo-checkbox-advertisement
1 year
The cookie is set by GDPR cookie consent to record the user consent for the cookies in the category "Advertisement".
cookielawinfo-checkbox-necessary
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookies is used to store the user consent for the cookies in the category "Necessary".
cookielawinfo-checkbox-performance
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Performance".
gdpr_status
6 months 2 days
This cookie is set by the provider Media.net. This cookie is used to check the status whether the user has accepted the cookie consent box. It also helps in not showing the cookie consent box upon re-entry to the website.
lang
This cookie is used to store the language preferences of a user to serve up content in that stored language the next time user visit the website.
viewed_cookie_policy
11 months
The cookie is set by the GDPR Cookie Consent plugin and is used to store whether or not user has consented to the use of cookies. It does not store any personal data.
Analytical cookies are used to understand how visitors interact with the website. These cookies help provide information on metrics the number of visitors, bounce rate, traffic source, etc.
Cookie
Duration
Description
SC_ANALYTICS_GLOBAL_COOKIE
10 years
This cookie is associated with Sitecore content and personalization. This cookie is used to identify the repeat visit from a single user. Sitecore will send a persistent session cookie to the web client.
vuid
2 years
This domain of this cookie is owned by Vimeo. This cookie is used by vimeo to collect tracking information. It sets a unique ID to embed videos to the website.
WMF-Last-Access
1 month 18 hours 24 minutes
This cookie is used to calculate unique devices accessing the website.
_ga
2 years
This cookie is installed by Google Analytics. The cookie is used to calculate visitor, session, campaign data and keep track of site usage for the site's analytics report. The cookies store information anonymously and assign a randomly generated number to identify unique visitors.
_gid
1 day
This cookie is installed by Google Analytics. The cookie is used to store information of how visitors use a website and helps in creating an analytics report of how the website is doing. The data collected including the number visitors, the source where they have come from, and the pages visted in an anonymous form.
Advertisement cookies are used to provide visitors with relevant ads and marketing campaigns. These cookies track visitors across websites and collect information to provide customized ads.
Cookie
Duration
Description
IDE
1 year 24 days
Used by Google DoubleClick and stores information about how the user uses the website and any other advertisement before visiting the website. This is used to present users with ads that are relevant to them according to the user profile.
test_cookie
15 minutes
This cookie is set by doubleclick.net. The purpose of the cookie is to determine if the user's browser supports cookies.
VISITOR_INFO1_LIVE
5 months 27 days
This cookie is set by Youtube. Used to track the information of the embedded YouTube videos on a website.
Performance cookies are used to understand and analyze the key performance indexes of the website which helps in delivering a better user experience for the visitors.
Cookie
Duration
Description
YSC
session
This cookies is set by Youtube and is used to track the views of embedded videos.
_gat_UA-62336821-1
1 minute
This is a pattern type cookie set by Google Analytics, where the pattern element on the name contains the unique identity number of the account or website it relates to. It appears to be a variation of the _gat cookie which is used to limit the amount of data recorded by Google on high traffic volume websites.