On October 3, 2022, the Nobel Assembly at Karolinska Institutet announced the 2022 Nobel Prize in Physiology or Medicine had been awarded to Svante Pääbo, director of the Department of Genetics at the Max Planck Institute for Evolutionary Anthropology in Leipzig, Germany. The Assembly cited his “discoveries concerning the genomes of extinct hominins and human evolution”. They mentioned the highlight of his research: the seemingly impossible task, at the time, of sequencing the Neanderthal genome. The discoveries that followed from this sequencing project continue to redefine our understanding of modern human origins.
The award showcases the technological advancements made in the analysis of ancient DNA. However, Pääbo’s research had an inauspicious beginning. In 1985, he published the results of his early work, cloning and sequencing DNA fragments from a 2,400-year-old Egyptian mummy (1). Unfortunately, later analysis revealed that the samples could have been contaminated by the researchers’ own DNA (2).
Imagine that you’re putting together a large, complex jigsaw puzzle, comprising thousands of exceptionally small pieces. You lay them all out and attempt to make sense of them. It would be far easier to assemble this puzzle were the pieces larger, containing more of the image advertised on the box. The same can be said when sequencing a genome.
Traditional short-read or next-generation sequencing relies on DNA spliced into small fragments (≤300 base pairs) and then amplified. While useful for detecting small genetic variants like single-base changes to the DNA, this type of sequencing can fail to illuminate larger variations (typically over 50 base pairs) in the genome. Long-read sequencing, or third generation sequencing, allows more accurate genome assemblies, facilitating better detection of structural variants like copy number variations, duplications, translocations and inversions that are too large to identify with short-read sequencing. Long-read sequencing has the capability to fill in “dark regions” of a genome that are unfinished and can be used to assemble larger, more complex genomes using longer fragments of DNA, or high-molecular weight (HMW) DNA.
Researchers looking for new chemistry for Sanger sequencing need look no further than the ProDye™ Terminator Sequencing System, developed by Promega for use in capillary electrophoresis instruments. Sanger sequencing, or dye-terminator sequencing, has been the gold standard of DNA analysis for over 40 years and is a method commonly used in labs around the world. Even as new technologies emerge, Sanger sequencing remains the most cost-effective method for sequencing shorter pieces of DNA.
The rapid advancement of next-generation sequencing technology, also known as massively parallel sequencing (MPS), has revolutionized many areas of applied research. One such area, the analysis of mitochondrial DNA (mtDNA) in forensic applications, has traditionally used another method—Sanger sequencing followed by capillary electrophoresis (CE).
Although MPS can provide a wealth of information, its initial adoption in forensic workflows continues to be slow. However, the barriers to adoption of the technology have been lowered in recent years, as exemplified by the number of abstracts discussing the use of MPS presented at the 29th International Symposium for Human Identification (ISHI 29), held in September 2018. Compared to Sanger sequencing, MPS can provide more data on minute variations in the human genome, particularly for the analysis of mtDNA and single-nucleotide polymorphisms (SNPs). It is especially powerful for analyzing mixture samples or those where the DNA is highly degraded, such as in human remains.
It is with sadness that we recognize the passing of Dr. Frederick Sanger. Sanger is known to molecular biologists and biochemists worldwide for his DNA sequencing technique, which won for him the 1980 Nobel prize in Chemistry.
Also noteworthy, Sanger’s laboratory accomplished the first complete genome sequence, that of a viral DNA genome more than 5,000 base pairs in length.
The 1980 prize was Sanger’s second Nobel award, his first awarded in 1958 for determining the chemical structure of proteins. In this work, Sanger elucidated not only the amino acids that comprised insulin but also the order in which the amino acids occurred.
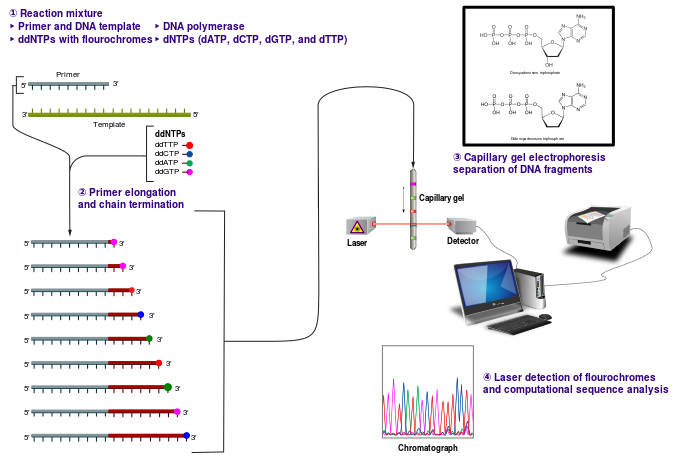
About Sanger Sequencing Sanger DNA sequencing is also known as the chain-termination method of sequencing. The Sanger technique uses dideoxynucleotides or ddNTPs in addition to typical deoxynucleotides (dNTPs) in the reaction. ddNTPs result in termination of the DNA strand because ddNTPs lack the 3’-OH group required for phosphodiester bond formation between nucleotides. Without this bond, the chain of nucleotides being formed is terminated.
Sanger sequencing requires a single-stranded DNA, a DNA primer (either radiolabeled or with a fluorescent tag), DNA polymerase, dNTPs and ddNTPs. Four reactions are set up, one for each nucleotide, G, A, T and C. In each reaction all four dNTPs are included, but only one ddNTP (ddATP, ddCTP, ddGTP or ddTTP) is added. The sequencing reactions are performed and the products denatured and separated by size using polyacrylamide gel electrophoresis.
Diagram of Sanger dideoxy sequencing. (Courtesy of Wikipedia and Estevez, J.)
This reaction mix results in various lengths of fragments representing, for instance, the location of each A nucleotide in the sequence, because while there is more dATP than ddATP in the reaction, there is enough ddATP that each ATP ultimately instead is replaced with a ddATP, resulting in chain termination. Separation by gel electrophoresis reveals the size of these ddATP-containing fragments, and thus the locations of all A nucleotide in the sequence. Similar information is provided for GTP, CTP and TTP.
The Maxam and Gilbert DNA sequencing method had the advantage at the time of being used with double-stranded DNA. However, this method required DNA strand separation or fractionation of the restriction enzyme fragments, resulting in a somewhat more time-consuming technique, compared to the 1977 method published by Sanger et al.
Dr. Sanger was born in Gloucestershire, U.K. in 1918, the son of a physician. Though he initially planned to follow his father into medicine, biochemistry became his life-long passion and area of research endeavor. Sanger retired at age 65, to spend more time at hobbies of gardening and boating.
DNA testing methods are being used to solve problems in an ever-increasing number of fields. From crime scene analysis to tissue typing, from mammoths to Neanderthals, and from Thutmose I to Richard III, both modern mysteries and age-old secrets are being revealed. The availability of fast, accurate, and convenient DNA amplification and sequencing methods has made DNA analysis a viable option for many types of investigation. Now it is even being applied to solve such mundane mysteries as the precise ingredients used in a sausage recipe, and to answer that most difficult of questions “what exactly is in a doner kebab?” Continue reading “Dietary Analysis, DNA Style”
For sixty years now, scientists have studied the role of DNA as a vehicle for the storage and transmission of genetic information from generation to generation. We have marveled at the capacity of DNA to store all the information required to describe a human being using only a 4-letter code, and to pack that information into a space the size of the nucleus of a single cell. A letter published last week in Nature exploits this phenomenal storage capacity of DNA to archive a quite different kind of information. Forget CDs, hard drives and chips, the sum of human knowledge can now be stored in synthetic DNA strands. The Nature letter, authored by scientists from the European Bioinformatics Institute in Cambridge, UK, and Agilent Technologies in California, describes a proof-of-concept experiment where synthetic DNA was used to encode Shakespeare’s Sonnets, Martin Luther King’s “I Have a Dream” speech, a picture of the Bioinformatics Institute, and the original Crick and Watson paper on the double-helical nature of DNA. Continue reading “Sonnets in DNA”
XWe use cookies and similar technologies to make our website work, run analytics, improve our website, and show you personalized content and advertising. Some of these cookies are essential for our website to work. For others, we won’t set them unless you accept them. To learn more about our approach to Privacy we invite you to Read More
By clicking “Accept All”, you consent to the use of ALL the cookies. However you may visit Cookie Settings to provide a controlled consent.
We use cookies and similar technologies to make our website work, run analytics, improve our website, and show you personalized content and advertising. Some of these cookies are essential for our website to work. For others, we won’t set them unless you accept them. To find out more about cookies and how to manage cookies, read our Cookie Policy.
If you are located in the EEA, the United Kingdom, or Switzerland, you can change your settings at any time by clicking Manage Cookie Consent in the footer of our website.
Necessary cookies are absolutely essential for the website to function properly. These cookies ensure basic functionalities and security features of the website, anonymously.
Cookie
Duration
Description
cookielawinfo-checbox-analytics
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Analytics".
cookielawinfo-checbox-functional
11 months
The cookie is set by GDPR cookie consent to record the user consent for the cookies in the category "Functional".
cookielawinfo-checbox-others
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Other.
cookielawinfo-checkbox-advertisement
1 year
The cookie is set by GDPR cookie consent to record the user consent for the cookies in the category "Advertisement".
cookielawinfo-checkbox-necessary
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookies is used to store the user consent for the cookies in the category "Necessary".
cookielawinfo-checkbox-performance
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Performance".
gdpr_status
6 months 2 days
This cookie is set by the provider Media.net. This cookie is used to check the status whether the user has accepted the cookie consent box. It also helps in not showing the cookie consent box upon re-entry to the website.
lang
This cookie is used to store the language preferences of a user to serve up content in that stored language the next time user visit the website.
viewed_cookie_policy
11 months
The cookie is set by the GDPR Cookie Consent plugin and is used to store whether or not user has consented to the use of cookies. It does not store any personal data.
Analytical cookies are used to understand how visitors interact with the website. These cookies help provide information on metrics the number of visitors, bounce rate, traffic source, etc.
Cookie
Duration
Description
SC_ANALYTICS_GLOBAL_COOKIE
10 years
This cookie is associated with Sitecore content and personalization. This cookie is used to identify the repeat visit from a single user. Sitecore will send a persistent session cookie to the web client.
vuid
2 years
This domain of this cookie is owned by Vimeo. This cookie is used by vimeo to collect tracking information. It sets a unique ID to embed videos to the website.
WMF-Last-Access
1 month 18 hours 24 minutes
This cookie is used to calculate unique devices accessing the website.
_ga
2 years
This cookie is installed by Google Analytics. The cookie is used to calculate visitor, session, campaign data and keep track of site usage for the site's analytics report. The cookies store information anonymously and assign a randomly generated number to identify unique visitors.
_gid
1 day
This cookie is installed by Google Analytics. The cookie is used to store information of how visitors use a website and helps in creating an analytics report of how the website is doing. The data collected including the number visitors, the source where they have come from, and the pages visted in an anonymous form.
Advertisement cookies are used to provide visitors with relevant ads and marketing campaigns. These cookies track visitors across websites and collect information to provide customized ads.
Cookie
Duration
Description
IDE
1 year 24 days
Used by Google DoubleClick and stores information about how the user uses the website and any other advertisement before visiting the website. This is used to present users with ads that are relevant to them according to the user profile.
test_cookie
15 minutes
This cookie is set by doubleclick.net. The purpose of the cookie is to determine if the user's browser supports cookies.
VISITOR_INFO1_LIVE
5 months 27 days
This cookie is set by Youtube. Used to track the information of the embedded YouTube videos on a website.
Performance cookies are used to understand and analyze the key performance indexes of the website which helps in delivering a better user experience for the visitors.
Cookie
Duration
Description
YSC
session
This cookies is set by Youtube and is used to track the views of embedded videos.
_gat_UA-62336821-1
1 minute
This is a pattern type cookie set by Google Analytics, where the pattern element on the name contains the unique identity number of the account or website it relates to. It appears to be a variation of the _gat cookie which is used to limit the amount of data recorded by Google on high traffic volume websites.