Proteomics, the analysis of the entire protein content of a living system, has become a vital part of life science research, and mass spectrometry (MS) is the method for analyzing proteins. MS analysis of protein content allows researchers to identify proteins, sequence them and determine the nature of post translational modifications.
Mass spectrometry allows characterization of molecules by converting them to ions so that they can be manipulated in electrical and magnetic fields. Basically a small sample (analyte) is ionized, usually to cations by loss of an electron. After ionization, the charged particles (ions) are separated by mass and charge; the separated particles are measured and data displayed as a mass spectrum. The mass spectrum is typically presented as a bar graph where each peak represents a single charged particle having a specific mass-to-charge (m/z) ratio. The height of the peak represents the relative abundance of the particle. The number and relative abundance of the ions reveal how different parts of the molecule relate to each other.
For the study of large, organic macromolecules, matrix associated laser desorption/ionization (MALDI) or tandem mass spec/collision induced dissociation (MS/MS) techniques are often used to generate the charged particles from the analyte. MS analysis brings sensitivity and specificity to proteome analysis. The technique has excellent resolution and is able to distinguish one ion from another, even when their m/z ratios are similar. Macromolecules are present in extremely different concentrations in the cells, and MS analysis can detect biomolecules across five logs of concentration.
Sample Preparation Quality
Obtaining the sensitivity and accuracy that makes MS analysis such a highly accurate and valuable technique requires sophisticated and well-maintained instrumentation and careful sample preparation. MS instruments can be negatively affected by compounds from biological samples, particularly salts and detergents used in sample preparation procedures. These compounds can cause loss of sensitivity of the mass spec analysis and great frustration for the researcher.
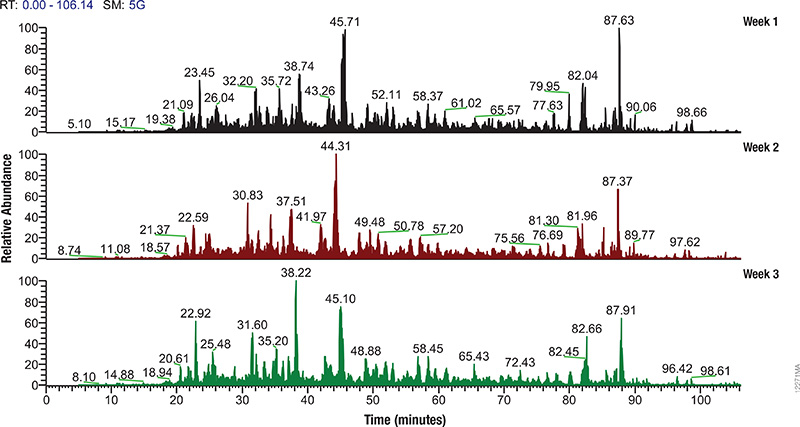
Mass Spec Instrument Performance
Regular monitoring of instrument performance and careful validation of sample preparation methods are essential to generating reliable, reproducible MS data. However such monitoring requires reference material of high complexity that generates reproducible results. Quite often typical reference extracts are produced in house, may come from commercial extracts that were generated for other purposes, or are left over from previous MS experiments. All of these extract sources are of variable quality and may not be the best for evaluating techniques and instruments.
The Mass Spec-Compatible Yeast and Human Protein Extracts from Promega provide complex protein reference material produced under tightly controlled conditions that allow researchers to evaluate MS instrument performance. Cell lysis of these samples has been optimized to allow maximum proteome recovery and produce minimal fragmentation. The digestion with Trypsin/Lys-C mix is exhaustive, and essentially all non-biological post-translational modifications such as deamidation, oxidation and carbamylation have been eliminated.
These extracts are provided as a ready-to-use, predigested format for applications such as assessing MS instrument performance or comparing lab to lab performance or as intact extracts for validating sample preparation.
In his recent webinar Monitoring Mass Spec Instrument Performance and Method Development with Whole Cell Human and Yeast Protein Reference Extracts, Dr. Sergei Saveliev discusses the features of theses extracts and the benefits of using them for a variety of MS applications in the laboratory.
Post updated 3/2/2021
Related Posts
Michele Arduengo
Latest posts by Michele Arduengo (see all)
- IL-6/STAT3-Regulated Long Non-Coding RNA Is Involved in Colorectal Cancer Progression - July 1, 2025
- Using Dual-Luciferase Assays to Identify the Role of Non-Coding RNAs in Disease - May 2, 2025
- An Unexpected Role for RNA Methylation in Mitosis Leads to New Understanding of Neurodevelopmental Disorders - March 27, 2025