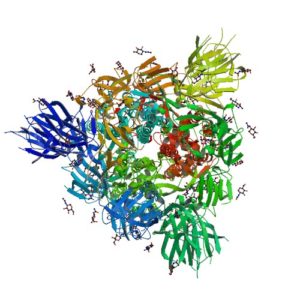
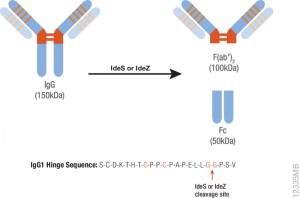
The spike protein of the SARS-CoV-2 virus is a very commonly researched target in COVID-19 vaccine and therapeutic studies because it is an integral part of host cell entry through interactions between the S1 subunit of the spike protein with the ACE2 protein on the target cell surface. Viral proteins important in host cell entry are typically highly glycosylated. Looking at the sequence of the SARS-CoV-2 virus, researchers predict that the spike protein is highly glycosylated. In a recent study, researchers conducted a glycosylation analysis of SARS-CoV-2 proteins using mass spec analysis to determine the N-glycosylation profile of the subunits that make up the spike protein.
Glycans assist in protein folding and help the virus avoid immune recognition by the host. Glycosylation can also have an impact on the antigenicity of the virus, as well as potential effects on vaccine safety and efficacy. Mass spectrometry is widely used for viral characterization studies of influenza viruses. Specifically, mass spec has been used to study influenza protein glycosylation, antigen quantification, and determination of vaccine potency.
Continue reading “Mass Spec for Glycosylation Analysis of SARS-CoV-2 Proteins Implicated in Host-Cell Entry”