This blog was written by guest contributor Tian Yang, Associate Product Manager, Promega, in collaboration with Kristin Huwiler, Manager, Small Molecule Drug Discovery, Promega.
Determining the selectivity of a compound is critical during chemical probe or drug development. In the case of chemical probes, having a clearly defined mechanism of action and specific on-target activity are needed for a chemical probe to be useful in delineating the function of a biological target of interest in cells. Similarly, optimizing a drug candidate for on-target potency and reducing off-target interactions is important in the drug development process (1,2). A thorough understanding of the selectivity profile of a drug can facilitate drug repurposing, by enabling approved therapeutics to be applied to new indications (3). Interestingly, small molecule drugs do not necessarily require the same selectivity as a chemical probe, since some drugs may benefit from polypharmacology to achieve their desired clinical outcome.
Selectivity profiling panels based on biochemical methods have commonly been used to assess compound specificity for established target classes in drug discovery and chemical probe development. Biochemical assays are target-specific and often quantitative, enabling direct measurements of compound affinities for targets of interest and facilitate comparison of compound engagement to a panel of targets. As an example, several providers offer kinase selectivity profiling services using different assay formats and kinase panels comprised of 100 to 400 kinases (4). However, just as biochemical target engagement does not always translate to cellular activity, selectivity profiles based on biochemical platforms may not reflect compound selectivity in live cells (5).
Cellular Selectivity Profiling Methods
With advancement in techniques for measuring cellular target engagement, compound selectivity can also be profiled in live cells, providing a more physiologically relevant measurement of the on- and off-target interactions of that compound. Here we review some of the most common cellular approaches that have been used.
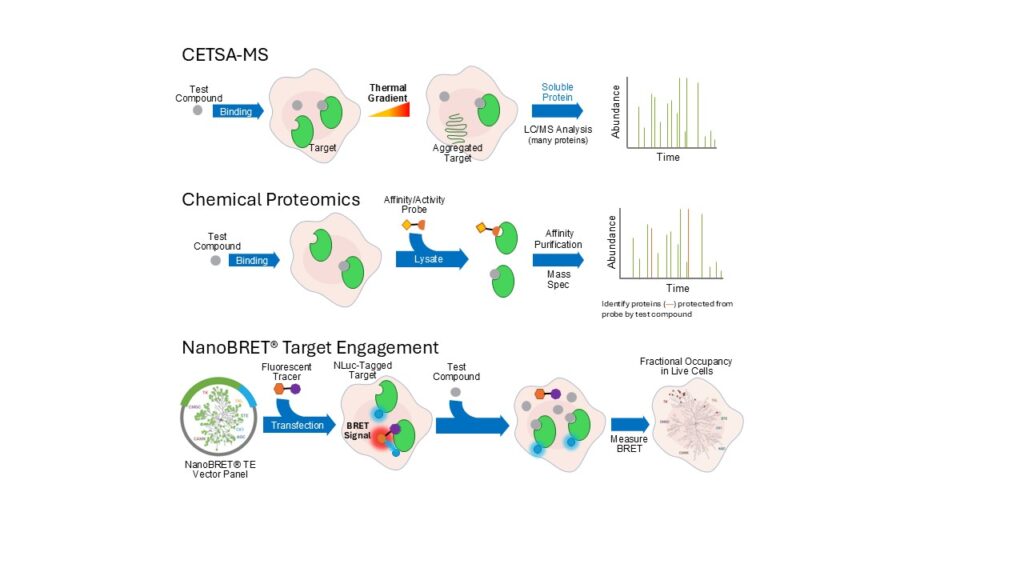
Chemical proteomics is well-suited to interrogate proteome-wide binding interactions. Using probes derived from compounds of interest, chemical proteomics can enrich and detect proteins bound to the probe directly. Competition of the probes by the parent compounds provides validation of the genuine protein targets for the compounds of interest, generating a proteome-wide profile of compound selectivity (6,7). In the absence of compound-specific probes, affinity- or activity-based probes based on promiscuous pharmacophores, can be used to assess compound selectivity among related protein families via competition with probes for target binding.
Initially chemical proteomics are predominantly performed with cell lysates to accommodate the bulky capture moieties in the probes. With the development of live-cell compatible probes, where a bioorthogonal reactive group replaces the bulky capture moiety, probe and compound binding can be done in intact cells, before the cells are lysed and a capture handle is coupled to the probe for subsequent enrichment and mass spectrometry analysis (6).
Cellular thermal shift assay (CETSA) is a probe-free technique for characterizing compound binding to target proteins in cells. It assesses the ability of a compound to stabilize a protein to thermal challenge by measuring the protein aggregation that occurs upon heating cells. The change in the amount of
non-aggregated protein, following heat treatment in the presence or absence of a compound, can be measured by lysing the cells and performing an immunoassay that recognizes a target of interest. CETSA can alternatively be coupled with mass spectrometry (CETSA-MS) to detect compound-induced changes in protein levels across the proteome (7). Although not all proteins will exhibit a shift in stability upon compound binding, CETSA-MS provides a more unbiased view of compound specificity in cells without the need for validated probe(s).
NanoBRET® Target Engagement (TE) Assays serve as an alternative to the mass spectrometry-based approaches for assessing cellular selectivity against a panel of related proteins. Leveraging bioluminescence resonance energy transfer (BRET) between NanoLuc®-tagged target proteins and target-binding fluorescent probes, NanoBRET® TE Assays directly and quantitatively measure apparent compound affinity and target occupancy via probe displacement without the need to lyse the cells. The quantitative affinity and occupancy measurements enable comparisons of the binding of many different compounds to one target, as well as engagement of one compound to many different targets. The use of probes with promiscuous binding profiles allows for wide target coverage with only a handful of probes, which, combined with an addition-only workflow, facilitates the development of high-throughput screening for selectivity profiling (8,9).
Cellular functional assays, such as receptor internalization, reporter gene and ion flux assays, have also been used to determine compound specificity among groups of related proteins in live cells (10-12). These assays measure the downstream effects of compound engagement to the targets. However, careful assay design and proper controls are needed to link results of these assays to on-target engagement in cells and reduce the likelihood of false-positives caused by nonspecific or off-target effects.
Uncovering Novel Compound-Target Interactions in Cells
Using proteomics-based cellular target engagement approaches provides a more comprehensive assessment of compound off-target interactions. In their 2016 publication, Becher and colleagues applied both chemical proteomics and CETSA-MS to identify undescribed off-targets for the FDA-approved HDAC inhibitor Panobinostat (13). In addition to identifying the HDAC proteins, which are the expected targets for this pan-HDAC inhibitor, the authors also identified the protein tetratricopeptide repeat domain 38 (TTC38) and phenylalanine hydroxylase (PAH) as off-targets for the drug. PAH plays a role in phenylalanine metabolism, and its inhibition by Panobinostat could explain the hypothyroidism-like symptoms commonly observed with Panobinostat treatment. On the other hand, PAH inhibition could provide a therapeutic opportunity for managing symptoms associated with type I tyrosinemia, and Panobinostat may be a candidate for drug repurposing or serve as a scaffold for developing selective PAH inhibitors (7,13).
Novel compound-target interactions can be identified when performing live-cell selectivity profiling against a panel of targets and comparing to the same panel of targets tested biochemically. When Sorafenib, a kinase inhibitor drug approved for treatment of hepatocellular carcinoma, renal cell carcinoma and differentiated thyroid carcinoma, was profiled against a panel of 192 kinases in live cells using the NanoBRET® TE platform, the overall selectivity of the drug improved in comparison to the selectivity profile obtained from cell-free biochemical analysis. This observation is not uncommon, as compound permeability and competition by cellular ATP are known to decrease kinase inhibitor affinity in live cells (14). More interestingly, cellular analysis revealed two off-targets, NTRK2 and RIPK2, for Sorafenib that were missed by biochemical profiling. RIPK2 has been identified as a therapeutic target or prognostic marker for a number of cancers, including renal cell carcinoma (15). While additional studies are needed to determine whether these two kinases play any role in the therapeutic efficacy and/or the adverse effects of Sorafenib, the results highlight the benefit of cellular selectivity profiling for de-risking compounds during drug discovery and for identifying new chemotypes for potential targets of interest.
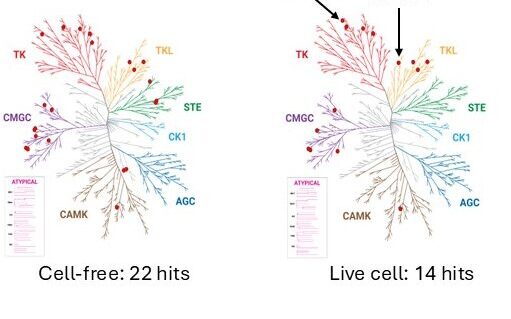
This figures compares kinases engaged (red dots) with >50% occupancy by 1µM Sorafenib against a panel of 192 kinases in cell-free and live-cell systems. Sorafenib displayed an improved selectivity profile in cells, but cellular profiling also revealed two new off-targets.
Summary
Selectivity is a cornerstone of successful drug discovery and chemical probe development, defining the precision with which a compound engages its intended target(s). While biochemical assays offer valuable insights into compound-target interactions, they often fail to predict true cellular selectivity, as the cellular environment significantly influences compound behavior. Recent advancements in cellular target engagement techniques—including chemical proteomics, CETSA-MS, and NanoBRET® Target Engagement assays—have enhanced our ability to profile compound selectivity directly within cells, providing more physiologically relevant data. These cellular assays can refine biochemical results and reveal novel off-target interactions previously undetectable by biochemical methods. As highlighted by the examples of Panobinostat and Sorafenib, cellular selectivity profiling represents a vital tool for uncovering novel compound-target interactions, de-risking drug candidates, and expanding the therapeutic potential of existing compounds.
References
- Arrowsmith, C.H. et al. (2015) The promise and peril of chemical probes. Nat. Chem. Biol. 11(8), 536-541.
- Huggins, D.J. et al. (2012) Rational Approaches to Improving Selectivity in Drug Design. J. Med. Chem. 55(4), 1424-1444.
- Palve, V. et al. (2021) Turning liabilities into opportunities: Off-target based drug repurposing in cancer. Semin. Cancer Biol. 68, 209-229.
- Uitdehaag, J.C.M. et al. (2012) A guide to picking the most selective kinase inhibitor tool compounds for pharmacological validation of drug targets. Br. J. Pharmacol. 166(3), 858-876.
- Kooijman, J.J. et al. (2022) Comparative kinase and cancer cell panel profiling of kinase inhibitors approved for clinical use from 2018 to 2020. Front. Oncol. 12, 953013.
- Robers, M.R. et al. (2020) Quantifying Target Occupancy of Small Molecules Within Living Cells. Annu. Rev. Biochem. 89, 557-581.
- Meissner, F. et al. (2022) The emerging role of mass spectrometry-based proteomics in drug discovery. Nat. Rev. Drug Discov. 21, 637-654.
- Nieman, A.N. et al. (2023) NanoBRET™ Live-Cell Kinase Selectivity Profiling Adapted for High-Throughput Screening. Methods Mol. Biol. 2706, 97-124.
- Schwalm M.P. and Knapp, S. (2025) Single-plate kinome screening in live-cells to enable highly cost-efficient kinase inhibitor profiling. SLAS Discov. 31, 100214.
- Addis, P. et al. (2024) Key aspects of modern GPCR drug discovery. SLAS Discov. 29, 1-22.
- McManus, O.B. et al. (2012) In: Markossian, S., Grossman, A., Arkin, M. et al., editors. Assay Guidance Manual [Internet]. Bethesda (MD): Eli Lilly & Company and the National Center for Advancing Translational Sciences; 2004-. Available from: https://www.ncbi.nlm.nih.gov/sites/books/NBK100915/
- Toporova, L. et al. (2020) Assessing the Selectivity of FXR, LXRs, CAR, and RORγ Pharmaceutical Ligands With Reporter Cell Lines. Front. Pharmacol. 11, 1122.
- Becher, I. et al. (2016) Thermal profiling reveals phenylalanine hydroxylase as an off-target of panobinostat. Nat. Chem. Biol. 12, 908-910.
- Schwaid, A.G. and Cornella-Taracido, I. (2017) Causes and Significance of Increased Compound Potency in Cellular or Physiological Contexts. J. Med. Chem. 61(5), 1767-1773.
- Rivoal, M. et al. (2023) Receptor Interacting Ser/Thr-Protein Kinase 2 as a New Therapeutic Target. J. Med. Chem. 66(21), 14391-14410.