Post-translational modifications of proteins are critical for proper protein function. Modifications such as phosphorylation/dephosphorylation can act as switches that activate or inactivate proteins in signaling cascades. The addition of specific sugars to membrane proteins on cells are critical for recognition, interaction with the extracellular matrix and other activities. While we know volumes about some types of protein modifications, ADP-ribosylation on aspartate and glutamate residues has been more difficult to study because of the chemical instability of these ester-linked modifications.
Matić Lab (Eduardo José Longarini and Ivan Matić) recently published a study that explored mono-ADP-ribosylation (ADPr) on aspartate and glutamate residues by the protein PARP1 and its potential reversal by PARG. PARP1 and PARG signaling are central to DNA repair and apoptosis pathways, making them potentially powerful therapeutic targets in cancer or neurodegenerative diseases in which DNA repair processes are often disrupted.
The spike protein of the SARS-CoV-2 virus is a very commonly researched target in COVID-19 vaccine and therapeutic studies because it is an integral part of host cell entry through interactions between the S1 subunit of the spike protein with the ACE2 protein on the target cell surface. Viral proteins important in host cell entry are typically highly glycosylated. Looking at the sequence of the SARS-CoV-2 virus, researchers predict that the spike protein is highly glycosylated. In a recent study, researchers conducted a glycosylation analysis of SARS-CoV-2 proteins using mass spec analysis to determine the N-glycosylation profile of the subunits that make up the spike protein.
Glycans assist in protein folding and help the virus avoid immune recognition by the host. Glycosylation can also have an impact on the antigenicity of the virus, as well as potential effects on vaccine safety and efficacy. Mass spectrometry is widely used for viral characterization studies of influenza viruses. Specifically, mass spec has been used to study influenza protein glycosylation, antigen quantification, and determination of vaccine potency.
The use of mass spectrometry for the characterization of individual or complex protein samples continues to be one of the fastest growing fields in the life science market.
Bottom-up proteomics is the traditional approach to address these questions. Optimization of each the individual steps (e.g. sample prep, digestion and instrument performance) is critical to the overall success of the entire experiment.
To address issues that may arise in your experimental design, Promega has developed unique tools and complementary webinars to help you along the way.
Here you can find a summary of individual webinars for the following topics:
Large-scale analyses of the proteome have revealed proteomic changes in response to disease, and these changes hold great promise for diagnostics and treatment of complex diseases if proteomic analysis can be brought into the clinical laboratory. Successful and reliable large-scale proteomics requires sample preparation workflows that are reproducible, reliable and show little variability. To bring proteomics into the clinical laboratory, standardized procedures and workflows for sample prep and analysis are required to generate valid, actionable results on a time scale useful for the clinic.
The two most common sample types analyzed for clinical proteomics are body fluids and tissue biopsies. To process these kinds of samples, there are two initial steps: tissue solubilization, followed by proteolytic digestion. Solubilization of solid tissues is the most labor-intensive and produces the most variable results.
The introduction of pressure cycling technology (PCT) using Barocycler instrumentation has greatly improved both tissue solubilization and digestion consistency. The PCT-based sample preparation protocols generally utilize urea as a lysis buffer for protein denaturing and solubilization. Urea has several drawbacks including inhibiting trypsin activity and introducing unwanted modifications like carbamylation.
Lucas and colleagues analyzed whether replacing urea with SDC would produce similar tissue digestion profiles and improve the PCT method.
Are you looking for proteases to use in your research? Explore our portfolio of proteases today.
SDC allowed the use of higher temperatures compared to urea, and hence the first step (lysis, reduction, and alkylation) was performed at 56 °C. The second digestion step in the Barocycler was optimized, and the third step was eliminated. To further reduce digestion time, they capitalized on Rapid Trypsin/Lys-C. Rapid Trypsin/Lys-C maintains robust activity at 70 °C, and allowed Barocycler digestion to be performed in a single step, completing digestion in 30 cycles (approximately 30 min) rather than 105 minutes, streamlining the protocol.
The data presented an improved conventional tissue PCT approach in a Barocycler by replacing urea and proteolytic enzymes with SDC, N-propanol, and modified commercially available enzymes that have higher optimum temperatures.
Try a sample of high-efficiency Trypsin Platinum today!
Visit our website for more on Trypsin Platinum, Mass Spectrometry Grade, with enhanced proteolytic efficiency and superior autoproteolytic resistance.
It’s time to analyze your protein and you are trying to decide where to begin. You are asking questions like: Which protease do I choose? How much enzyme should I use in my digest? How long should I perform my digest?
Unfortunately, there is no one-size fits all answer to this type of question other than… “well it depends.” All protease digests will be a balance between denaturing the protein sample to allow access to cleavage sites, optimizing conditions for the protease to function, and compatibility with your workflow and downstream applications. We provide general guidelines that work for most samples, but frequently you will need to optimize the conditions need for your specific sample and application.
Here, I use the example of a trypsin digest for downstream mass spectrometry to highlight key questions to ask and factors that can be optimized for any digest.
While many proteases are used in bottom-up mass spectrometric (MS) analysis, trypsin (4,5) is the de facto protease of choice for most applications. There are several reasons for this: Trypsin is highly efficient, active, and specific. Tryptic peptides produced after proteolysis are ideally suited, in terms of both size (350–1,600 Daltons) and charge (+2 to +4), for MS analysis. One significant drawback to trypsin digestion is the long sample preparation times, which typically range from 4 hours to overnight for most protocols. Achieving efficient digestion usually requires that protein substrates first be unfolded either with surfactants or denaturants such as urea or guanidine. These chemical additives can have negative effects, including protein modification, inhibition of trypsin or incompatibility with downstream LC-MS/MS. Accordingly, additional steps are typically required to remove these compounds prior to analysis.
Brachylophosaurus was a mid-sized member of the hadrosaurid family of dinosaurs living about 78 million years ago, and is known from several skeletons and bonebed material from the Judith River Formation of Montana and the Oldman Formation of Alberta. Recent fossil evidence indicates structures similar to blood vessels in location and morphology, have been recovered after demineralization of multiple dinosaur cortical bone fragments from multiple specimens, some of which are as old as 80 Ma. These structures were hypothesized to be either endogenous to the bone (i.e., of vascular origin) or the result of biofilm colonizing the empty network after degradation of original organic components (i.e., bacterial, slime mold or fungal in origin). Cleland et al. (1) tested the hypothesis that these structures are endogenous and thus retain proteins in common with extant archosaur blood vessels that can be detected with high-resolution mass spectrometry and confirmed by immunofluorescence.
Asp-N, Sequencing Grade, is an endoproteinase that hydrolyzes peptide bonds on the N-terminal side of aspartic and cysteic acid residues: Asp and Cys. Asp-N activity is optimal in the pH range of 4.0–9.0. This sequencing grade enzyme can be used alone or in combination with trypsin or other proteases to produce protein digests for peptide mapping applications or protein identification by peptide mass fingerprinting or MS/MS spectral matching. It is suitable for in-solution or in-gel digestion reactions.
The following references illustrate the use of Asp-N in recent publications:
Protein sequence coverage
Jakobsson, M et al. (2013) Identification and characterization of a novel Human Methyltransferase modulating Hsp70 protein function through lysine methylation. J. Biol. Chem. 288, 27752–63.
Carroll, J. et. al. (2013) Post-translational modifications near the quinone binding site of mammalian complex I. J. Biol. Chem. 288, 24799–08.
Glycoprotein analysis
Siguier, B. et al. (2014) First structural insights into α-L-Arabinofuranosidases from the two GH62 Glycoside hydrolase subfamilies. J. Biol. Chem. 289, 5261–73.
Vakhrushev, S. et al. (2013) Enhanced mass spectrometric mapping of the human GalNAc-type O-glycoproteome with SimpleCells. Mol. Cell. Prot.12, 932–44.
Berk, J. et al. (2013) . O-Linked β-N- Acetylglucosamine (O-GlcNAc) Regulates emerin binding to autointegration Factor (BAF) in a chromatin and Lamin B-enriched “Niche”. J. Biol. Chem. 288, 30192–09.
Phosphoprotein analysis
Roux, P. and Thibault, P. (2013) The Coming of Age of phosphoproteomics –from Large Data sets to Inference of protein Functions. Mol. Cell. Prot.12, 3453–64.
Are you looking for proteases to use in your research? Explore our portfolio of proteases today.
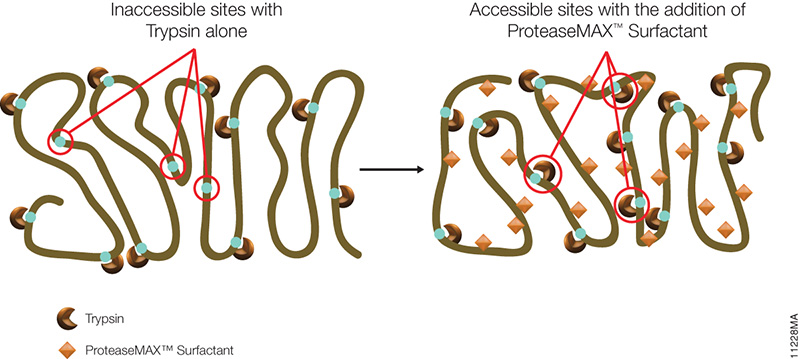
The novel mass spectrometry compatible surfactant sulfonate-(sodium 3-((1-(furan-2-yl)undecyloxy) carbonylamino)-propane-1-sulfonate (i.e.ProteaseMAX) facilitates both in-gel and in-solution digestion applications by reducing the time required, enabling protein solubilization/denaturation and increasing peptide/protein identifications.
A new application was highlighted in a recent publication (1) which utilized ProteaseMAX to lyse cells prior to trypsin digestion and subsequent mass spec analysis. The composition of the buffer determines the overall efficiency of cell lysis, dissociation of protein complexes, protein solubility and ease of removal prior to LC/MS-MS analysis.
Are you looking for proteases to use in your research? Explore our portfolio of proteases today.
When compared to lysis buffers containing either urea or SDC, ProteaseMAX provided the optimal number of identified peptides/proteins. In addition it can be easily removed from the lysate by acidic precipitation.
Reference
Pirmoradian, M. et al. (2013). Rapid and deep human proteome analysis by single-dimension shotgun proteomics. Mol. Cell. Prot.12, 3330–8.
XWe use cookies and similar technologies to make our website work, run analytics, improve our website, and show you personalized content and advertising. Some of these cookies are essential for our website to work. For others, we won’t set them unless you accept them. To learn more about our approach to Privacy we invite you to Read More
By clicking “Accept All”, you consent to the use of ALL the cookies. However you may visit Cookie Settings to provide a controlled consent.
We use cookies and similar technologies to make our website work, run analytics, improve our website, and show you personalized content and advertising. Some of these cookies are essential for our website to work. For others, we won’t set them unless you accept them. To find out more about cookies and how to manage cookies, read our Cookie Policy.
If you are located in the EEA, the United Kingdom, or Switzerland, you can change your settings at any time by clicking Manage Cookie Consent in the footer of our website.
Necessary cookies are absolutely essential for the website to function properly. These cookies ensure basic functionalities and security features of the website, anonymously.
Cookie
Duration
Description
cookielawinfo-checbox-analytics
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Analytics".
cookielawinfo-checbox-functional
11 months
The cookie is set by GDPR cookie consent to record the user consent for the cookies in the category "Functional".
cookielawinfo-checbox-others
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Other.
cookielawinfo-checkbox-advertisement
1 year
The cookie is set by GDPR cookie consent to record the user consent for the cookies in the category "Advertisement".
cookielawinfo-checkbox-necessary
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookies is used to store the user consent for the cookies in the category "Necessary".
cookielawinfo-checkbox-performance
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Performance".
gdpr_status
6 months 2 days
This cookie is set by the provider Media.net. This cookie is used to check the status whether the user has accepted the cookie consent box. It also helps in not showing the cookie consent box upon re-entry to the website.
lang
This cookie is used to store the language preferences of a user to serve up content in that stored language the next time user visit the website.
viewed_cookie_policy
11 months
The cookie is set by the GDPR Cookie Consent plugin and is used to store whether or not user has consented to the use of cookies. It does not store any personal data.
Analytical cookies are used to understand how visitors interact with the website. These cookies help provide information on metrics the number of visitors, bounce rate, traffic source, etc.
Cookie
Duration
Description
SC_ANALYTICS_GLOBAL_COOKIE
10 years
This cookie is associated with Sitecore content and personalization. This cookie is used to identify the repeat visit from a single user. Sitecore will send a persistent session cookie to the web client.
vuid
2 years
This domain of this cookie is owned by Vimeo. This cookie is used by vimeo to collect tracking information. It sets a unique ID to embed videos to the website.
WMF-Last-Access
1 month 18 hours 24 minutes
This cookie is used to calculate unique devices accessing the website.
_ga
2 years
This cookie is installed by Google Analytics. The cookie is used to calculate visitor, session, campaign data and keep track of site usage for the site's analytics report. The cookies store information anonymously and assign a randomly generated number to identify unique visitors.
_gid
1 day
This cookie is installed by Google Analytics. The cookie is used to store information of how visitors use a website and helps in creating an analytics report of how the website is doing. The data collected including the number visitors, the source where they have come from, and the pages visted in an anonymous form.
Advertisement cookies are used to provide visitors with relevant ads and marketing campaigns. These cookies track visitors across websites and collect information to provide customized ads.
Cookie
Duration
Description
IDE
1 year 24 days
Used by Google DoubleClick and stores information about how the user uses the website and any other advertisement before visiting the website. This is used to present users with ads that are relevant to them according to the user profile.
test_cookie
15 minutes
This cookie is set by doubleclick.net. The purpose of the cookie is to determine if the user's browser supports cookies.
VISITOR_INFO1_LIVE
5 months 27 days
This cookie is set by Youtube. Used to track the information of the embedded YouTube videos on a website.
Performance cookies are used to understand and analyze the key performance indexes of the website which helps in delivering a better user experience for the visitors.
Cookie
Duration
Description
YSC
session
This cookies is set by Youtube and is used to track the views of embedded videos.
_gat_UA-62336821-1
1 minute
This is a pattern type cookie set by Google Analytics, where the pattern element on the name contains the unique identity number of the account or website it relates to. It appears to be a variation of the _gat cookie which is used to limit the amount of data recorded by Google on high traffic volume websites.