For cancers that have proven challenging to target with traditional therapies, one emerging option is an approach called synthetic lethality. Synthetic lethality arises when inactivation of two gene products together lead to cell death but where inactivation of one does not (1, 2). Targeting a gene that is synthetic lethal to a cancer-related mutation creates an opening to specifically kill cancer cells while leaving healthy cells untouched.
In a recent study in Nature, scientists found that cells with amplification of CCNE1 are sensitive to inhibition of PKYMT1 kinase and identified a small molecule that is a selective inhibitor of PKYMT1 (3). When mice with tumor xenografts derived from CCNE1-high cell lines were dosed with the drug, researchers observed significantly slower tumor growth, and in some cases where the drug was co-dosed with another chemotherapeutic, tumor growth was completely halted.
Amplification of CCNE1 has been identified in about 20% of ovarian cancers and other difficult-to-treat cancers. CCNE1, however, codes for cyclin E, which “is not considered to be a druggable target” according to the study’s authors. Based on the researchers’ promising results for targeting CCNE1-high cells through synthetic lethal PKYMT1 inhibition, two different clinical trials have been initiated for adults with solid tumors.
Identifying PKMYT1 as an Essential Kinase in CCNE1-High Cells
The researchers initiated the study by identifying genes that are essential for cells that overexpress cyclin E. After creating a “CCNE1-high” cell line that stably overexpresses cyclin E, they carried out a loss of fitness CRISPR-Cas9 screen using a single guide RNA library in parent and CCNE1-high cell lines. From the screen, they were able to identify five genes that selectively caused a loss of fitness in CCNE1-high cells. Of the five genes, PKMYT1 showed the strongest “cancer dependency” in CCNE1-amplified tumor cell lines.
PKMYT1 encodes a protein kinase which negatively regulates CDK1 by phosphorylating CDK1 at Thr14 and sequestering CDK1 in the cytoplasm. PKMYT1 is structurally similar to the more well studied WEE1, which phosphorylates both CDK1 and CDK2. WEE1, however, did not appear in the researchers’ screen.
To confirm their initial screening results, the researchers knocked out PKMYT1 with targeted sgRNAs in CCNE1-high cells and the corresponding parent cells. Lethality was only observed for CCNE1-high cells and did not reduce parental cells’ viability. sgRNA targeting of WEE1 was lethal for all examined cell lines. Based on additional screens and the unique roles of PKMYT1 and WEE1, the researchers proposed a hypothesis where activation of CDK1 via inhibition of PKMYT1 leads to lethality in cells overexpressing CCNE1.
Determining Enzymatic Inhibition and Intracellular Target Engagement for a Selective Inhibitor of PKMYT1
Based on a screen of known kinase inhibitors and subsequent structure-guided medicinal chemistry efforts, the team identified azaindole derivative RP-6306 as a selective inhibitor of PKMYT1. For this drug to have utility in CCNE1-driven tumors, the authors needed to first prove that the drug inhibited PKMYT1 kinase activity and that the drug could permeate live cells and engage the target. Furthermore, the authors needed to evaluate the selectivity of RP-6306 against kinases similar to PKMYT1, to ensure that the drug effect was driven through the intended target. This can be notoriously challenging for kinase inhibitors, based on the highly conserved nature of the drug binding pocket, as well as the high concentration of intracellular ATP (4). Toward these goals, the authors combined biochemical and cellular assays to evaluate PKMYT1 target engagement.
An ADP-Glo™ kinase assay was used to determine the inhibitory effect of RP-6306 on the catalytic activity of recombinant PKMYT1. While RP-6306 showed an IC50 of 3.1±1.2nM toward PKMYT1 a related small molecule control showed, as expected, no effect on PKMYT1 activity in the assay.
RP-6306 was also found to selectively inhibit PKYMT1 relative to WEE1. Using a NanoBRET™ target engagement assay, the researchers determined the half maximal effective concentration of RP-6306 with a NanoLuc-PKMYT1 fusion protein was 2.5±0.8nM, which was 1,920-fold lower than the EC50 for target engagement of RP-6306 with a NanoLuc-WEE1 fusion (EC50 = 2.5±0.8µM). Additional studies showed that RP-630 treatment in CCNE1-high cells led to CDK1 kinase activation without CDK2 kinase activation.
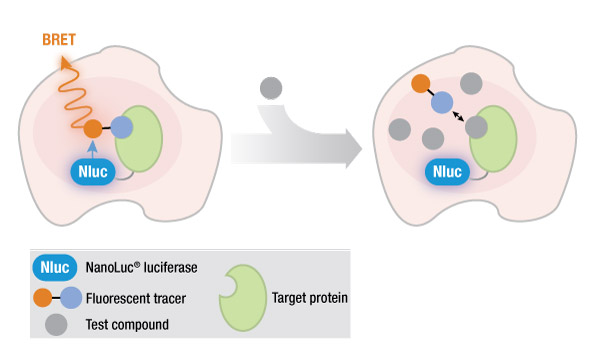
These studies demonstrated that the pharmacological activity of RP-6306 can selectively recapitulate the lethal effects of PKMYT1 knockout in CCNE1-high cells.
Read this application note to learn more about carrying out a NanoBRET™ TE assay with PKMYT1.
PKMYT1 Inhibitor RP-6306 Elicits Activity in Tumor Cells
Following further characterization of the effects of PKYMT1 inhibition in CCNE1-high cells and the pathways involved, the researchers examined the activity of RP-6306 against tumor xenografts in mice. For the implanted CCNE1-amplified cell lines HCC1569 and OVCAR3, oral dosing with RP-6306 led to tumor growth inhibition up to 79% and 84%, respectively. Conversely, the growth of tumors derived from CCNE1-normal cell lines SUM149PT and A2780 were not affected by RP-6306 dosing.
RP-6306 was also tested against a tumor xenograft derived from a human pancreatic adenocarcinoma, which displayed moderate CCNE1 amplification. Dosing with RP-6306 led to tumor growth inhibition of 64% over 48 days.
In these RP-6306 dosing experiments, the mouse models showed less than 7% body weight loss, indicating that the drug was well tolerated.
When they dosed mice with both RP-6306 and gemcitabine, a known chemotherapeutic agent, researchers observed significant regression in HCC1569- and OVCAR3-derived tumors. After a 21-day dosing period, tumors showed no sign of regrowth 15 days later. For the HCC1569 mouse models, 2 of the 7 mice were tumor-free. For the OVCAR3 models, 3 of 7 were tumor-free.
Based on these results, Repare Therapeutics, which partially funded the work, has launched two clinical trials on the efficacy of RP-6306 alone and in combination with gemcitabine for patients with solid tumors.
Interested in learning more kinase biology and drug discovery? Visit our applications webpage on tools for studying kinase biology for drug discovery applications.
References
- Mullard, A. (2022) What’s Next for the Synthetic Lethality Drug Discovery Engine? Nat. Rev. Drug Dis. 21, 477–479. doi: 10.1038/d41573-022-00107-0
- Kaelin, W. G., Jr. (2005) The Concept of Synthetic Lethality in the Context of Anticancer Therapy. Nat. Rev. Cancer 5, 689–698. doi: 10.1038/nrc1691
- Gallo, D., et al. (2022) CCNE1 Amplification is Synthetic Lethal with PKYMT1 Kinase Inhibition. Nature 604, 749–766. doi: 10.1038/s41586-022-04638-9
- Vasta, J. D., et al. (2018) Quantitative, Wide-Spectrum Kinase Profiling in Live Cells for Assessing the Effect of Cellular ATP on Target Engagement. Cell Chem. Biol. 25, 206–214.e11 doi: 10.1016/j.chembiol.2017.10.010
Latest posts by Jordan Nutting (see all)
- The Central Dogma of Promega: The Story and Science Behind Our Kit Packaging Design - May 7, 2024
- Silencing the Immunogenicity of AAV Vectors - April 4, 2024
- Discovering Cyclic Peptides with a “One-Pot” Synthesis and Screening Method - February 29, 2024