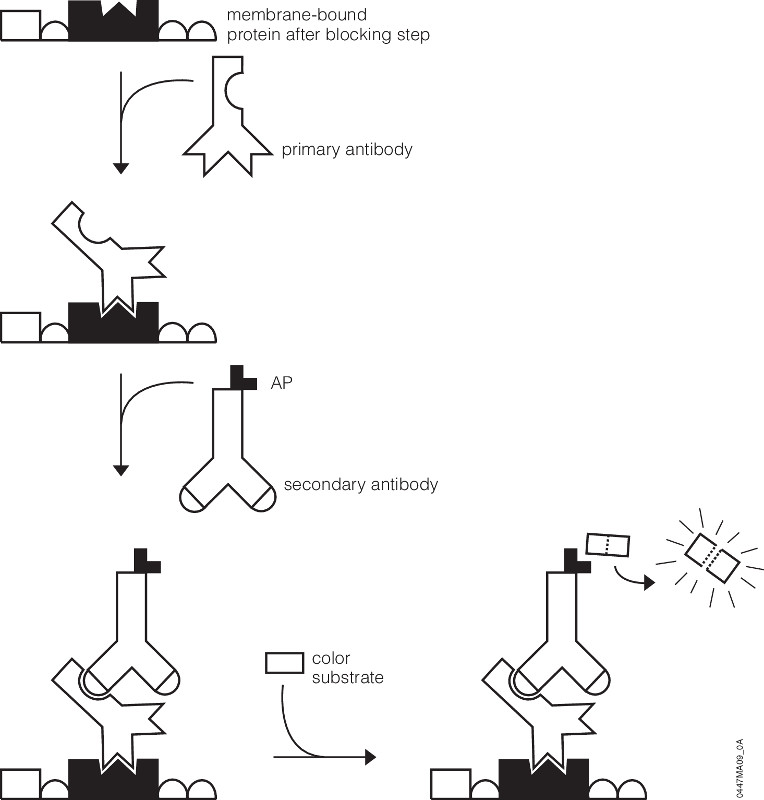
You’ve probably been there. You’ve got a new antibody or you’re testing out one you’ve made yourself. After weeks or months of work, your antibody is going to help move your research project forward. As you excitedly head to the dark room to develop your film, your mood is crushed when you see…bands, more bands, and smears. Alas, science has played one more cruel joke on you as you experience what so many of your fellow scientists have before. Despite such a dismal beginning, you often can still get good western blots by changing steps in your protocol.
Several steps in the western blot protocol can be optimized.
You may be using too much sample, the protein you’re trying to detect may not be stable in the lysis buffer you use or it degrades rapidly. In these situations you might try adding less lysate or using a different lysis buffer, and adding protease inhibitors. Most other optimizations for a western blot can be tried once you’ve got you’ve transferred from your gel to your blot (typically PVDF or nitrocellulose). Optimizing the blocking step can often make a huge difference in reducing background. Although 5% non-fat dry milk, 3% BSA, or normal serum are all commonly used blocking agents, some western blot assays perform much better with only one of these. Switching the blocking agent can sometimes improve performance. In particular, if you use a phospho-specific antibody you will want to block with BSA because 5% non-fat dry milk usually gives very high background due to the presence of phosphoproteins (casein). The length of blocking time may also need to be increased if you see excessive background. If your developed film has a high background but you see gaps between lanes with lysate, or empty lanes show no background, this indicates you’ve washed the blot sufficiently and you need to further optimize the antibody dilutions. If you see high background even in empty lanes, you need to perform additional washes prior to detection. Sub-optimal dilutions of primary and secondary antibodies will also give high background due to non-specific binding of proteins in your lysate from the primary antibody, secondary antibody or from both antibodies. Typically, primary antibodies are diluted 1:500 to 1:2000 and secondaries from 1:5000 to 1:20,000. If you’re seeing especially high background, try further diluting your antibodies. A quick method to determine the optimal sample concentration and antibody concentrations is a dot blot. You can start by taking a strip of a membrane (1 cm x 10 cm is a good starting range) and make serial dilutions of your lysate (5–10 dilutions) and place spots of lysate on the strip. Be sure to mark where you’re adding your sample (a pencil comes in handy here). Repeat with several more replicate strips. On each of these strips you can then test a different dilution of your primary or secondary antibodies. By creating a matrix of dilutions, you can quickly determine which primary and secondary dilutions yield the optimal conditions for your sample. You will also want to run a negative control where you only spot lysate and probe with primary or secondary, or where you spot no lysate and probe with primary and secondary. This will also tell you if your antibodies are causing high background. Other causes of high background include exposure times that are too long. For a really good antibody, you may only need a half-minute to several minute exposure to your film. Other antibodies that are less sensitive may require 30 minutes or more. If you’ve have a really sensitive antibody, decrease the exposure time. Your experiment may require additional optimizations, but generally the tips above are the best starting points and work for most westerns blots.
Ben Schmidt
Latest posts by Ben Schmidt (see all)
- Tips for Successful Dual-Reporter Assays - December 6, 2019
- Why You Don’t Need to Select a Wavelength for a Luciferase Assay - June 7, 2019
- To inject or not inject? - September 15, 2014