This blog was written by guest contributor Tian Yang, Associate Product Manager, Promega, in collaboration with Kristin Huwiler, Manager, Small Molecule Drug Discovery, Promega.
For target-based drug discovery programs, biochemical assays using purified target proteins are often run for initial hit discovery, as these assays are target-specific, quantitative and amenable for high-throughput screens, allowing for precise characterization of target-compound interactions. However, proteins do not act in isolation inside the cells. Instead, proteins form complexes with other cellular components to drive cellular processes, signaling cascades, and metabolic pathways. Just as the interactions between a target protein and its binding partners can influence the target function, compound engagement with target proteins can vary depending on the protein complex formed.
Protein-Protein Interactions can Impact Compound Binding Affinity and Duration
Protein-protein interactions (PPIs) are fundamental to all cellular processes, and therefore it is unsurprising that PPIs can also affect compound-protein interactions. For example, Cyclin-Dependent Kinases (CDKs) bind to specific cyclins or regulatory partners, which modulate the activities of the CDKs. Several CDKs can complex with multiple cyclins, allowing for dynamic regulation of the CDK activities (1). In a 2020 publication, the scientists at Promega and the Structural Genomics Consortium assessed the binding profile of known CDK inhibitors across the CDK family in live cells. They found that compound binding affinity can change drastically when target CDKs were co-expressed with specific cyclin partners (2). In particular, the authors showed that, for four CDK inhibitors representing different chemotypes, Dinaciclib and RGB-286638 preferentially engaged the CDK2-Cyclin E1 complex, whereas NVP-LCQ-195 and SB-1317 displayed similar affinity for both CDK2-Cyclin E1 and CDK2-Cyclin A1 complexes, demonstrating the impact of PPIs on compound engagement.
PPIs can also create new binding pockets for small molecules. In an effort to elucidate the mechanism of action for Trametinib, a clinical MEK kinase inhibitor, Khan and colleagues obtained co-crystal structures of Trametinib bound to MEK in complex with the scaffold protein KSR (3). Structural analysis showed that, compared to other allosteric MEK inhibitors, Trametinib uniquely bound to the interface of the MEK-KSR complex and formed contact with both proteins. Subsequent analysis by both cellular target engagement assays and in vitro binding assays revealed that binding to MEK-KSR complex enhanced the residence time of Trametinib.
Case Study: Targeting the PRMT5-MTA Complex in Precision Medicine
Beyond PPIs, proteins can interact with other macromolecules like nucleic acids and lipids or cellular metabolites, which can also influence compound-protein interactions. Protein arginine methyltransferase 5 (PRMT5) serves as an interesting example of how protein-metabolite complexes impact compound binding and how this impact can be leveraged in drug development.
PRMT5 catalyzes the transfer of a methyl group from the co-factor S-adenosylmethionine (SAM) to the arginine side chains of a variety of substrates that are involved in various cellular processes, such as cell signaling and differentiation, RNA splicing and DNA damage response, making PRMT5 essential to cell survival (4). The metabolite 5-methylthioadenosine (MTA) is an analog of SAM and can selectively inhibit the activity of PRMT5 by competitively displacing SAM. The cellular level of MTA is kept low by the enzyme 5’-methylthioadenosine phosphorylase (MTAP), which breaks down MTA as part of the methionine salvage pathway, preventing MTA from interfering with PRMT5 activity (5).
In ~10-15% of cancers, the MTAP gene is homozygously deleted, and the loss of MTAP expression leads to an accumulation of MTA inside the cells. Elevated MTA partially inhibits PRMT5 and sensitizes MTAP-deficient cancer cells to further inhibition of PRMT5 (4,5), creating a vulnerability in MTAP-deficient cancer cells and an opportunity for targeting these cells with precision medicine. However, the first generation of PRMT5 inhibitors, while displaying good selectivity and activity against PRMT5, inhibits PRMT5 in both healthy cells and MTAP-deficient cancer cells without distinction, causing toxicity and limiting therapeutic potential (4). To exploit the vulnerability of PRMT5 in MTAP-deficient cancers, PRMT5 inhibitors that preferentially engage the PRMT5-MTA complex are needed, as these inhibitors would selectively target cells with high MTA levels.
To develop PRMT5 inhibitors selective for MTAP-deficient cancer cells, a variety of biochemical assays were used for hit identification and lead optimization, including surface plasmon resonance, fluorescence anisotropy, and enzyme activity assays such as MTase-Glo™ Methyltransferase Assay. Regardless of the method, assays were performed in the presence of MTA to select compounds that bind the PRMT5-MTA protein-metabolite complex (6-8). PRMT5 without MTA or in complex with SAM was used as counter screens to identify and optimize leads for improved selectivity for the PRMT5-MTA complex, maximizing the preferential engagement to PRMT5 in MTAP-deficient cancers. To confirm cellular activity and selectivity, lead compounds are assayed in an isogenic pair of MTAP wildtype and MTAP-deficient cell lines and assessed for preferential inhibition of PRMT5-dependent methylation and cytotoxicity in MTAP-deficient cell lines. These efforts led to the development of a new class of MTA-cooperative PRMT5 inhibitors, some of which are now in clinical trials and have shown much improved safety profile (4).
Directly Characterize Compound Binding to PRMT5-MTA Complex in Live Cells
With emerging techniques to measure cellular target engagement, it is possible to quantitatively and directly characterize the binding of PRMT5 inhibitors to the PRMT5-MTA complex in cells. Specifically, the NanoBRET® Target Engagement (TE) platform has been applied to PRMT5 and enables the assessment of the first-generation of PRMT5 inhibitors, which were not designed to target the PRMT5-MTA complex, as well as the new MTA-cooperative PRMT5 inhibitors. To bias PRMT5 towards the formation of the PRMT5-MTA complex, exogenous MTA, which is cell permeable, can be added directly to cells to increase the intracellular MTA level. Similar to biochemical assays, MTA-cooperative PRMT5 inhibitors can be identified by comparing the compound binding affinity in the presence and absence of exogenous MTA. The NanoBRET® TE PRMT5 Assay can also be used in MTAP-deficient cells, quantifying the PRMT5 inhibitor affinity in a disease-relevant model. By directly measuring compound binding to PRMT5, the NanoBRET® TE assay links the results of cellular functional assays to compound target engagement in live cells.
PRMT5 Engagement by MTA-cooperative Inhibitors
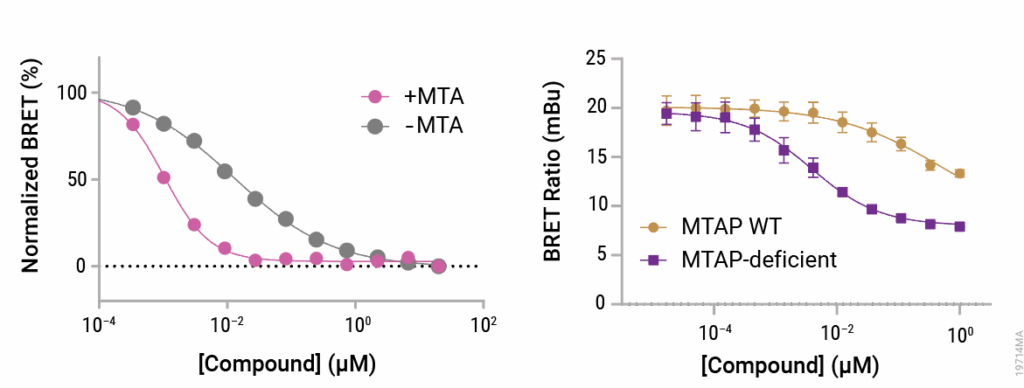
Figure 1. Binding to PRMT5 by MTA-cooperative inhibitor was assayed by NanoBRET® TE assay.
Left panel: Assay was performed in HEK293 cells, and intracellular MTA level was increased by adding exogenous MTA. Right panel: Assay was performed in an isogenic pair of MTAP wildtype (WT) and MTAP-deficient cell lines.
Imaging PRMT5-MTA Complex Engagement in Live Cells
In addition to quantifying compound affinity through the NanoBRET® TE PRMT5 Assay, engagement of PRMT5 within the context of the PRMT5-MTA complex can be visualized directly using the GloMax® Galaxy Bioluminescence Imager. MRTX1719, a selective PRMT5 inhibitor designed to exploit the metabolic vulnerability of MTAP-deleted cancers, demonstrates enhanced displacement of the NanoBRET® tracer in the presence of MTA—consistent with cooperative binding to the PRMT5-MTA complex (8). These imaging results reinforced prior biochemical findings and complemented the cell-based assays by offering insights into target engagement at the single-cell level.
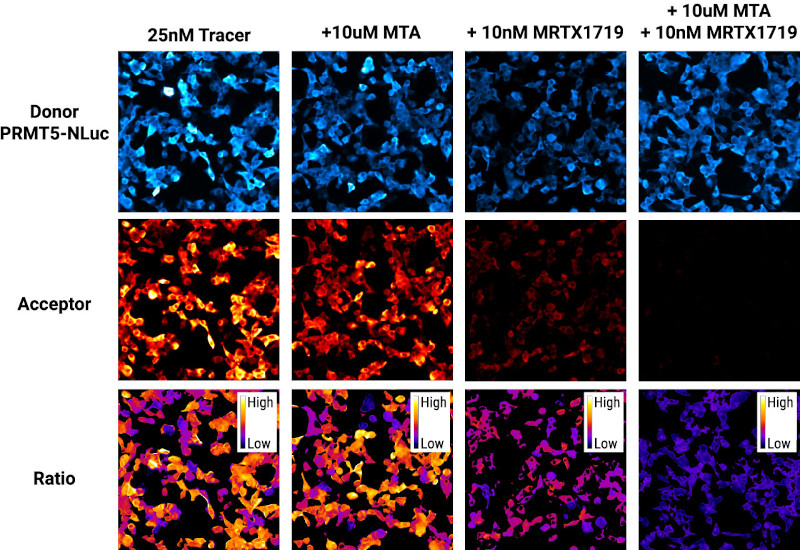
Figure 2. Visualizing synergistic effects of PRMT5 inhibitors with the GloMax® Galaxy Bioluminescence Imager. Top row: bioluminescence images of PRMT5-NanoLuc® donor captured with 3-minute exposures. Middle row: Acceptor images of the NanoBRET® tracer captured with a 3-minute exposure.
Bottom row: Ratiometric images generated by dividing the Acceptor signal by the Donor signal, scaled to milliBRET units. All images were pseudocolored during post-processing using Fiji (ImageJ).
Conclusion
Measuring compound-target engagement within biologically relevant protein complexes provides a deeper and more accurate understanding of how small molecules interact with their targets, as interactions between the target and other cellular components can significantly alter compound binding affinity, residence time, and ultimately, efficacy. The case of PRMT5 and the development of MTA-cooperative inhibitors underscores the power of leveraging complex-specific vulnerabilities for precision medicine. By designing hit finding campaigns around the PRMT5-MTA complex, researchers identified compounds with improved selectivity and safety profile for targeting MTAP-deficient cancers. This approach not only improved the therapeutic potential of PRMT5 inhibitors but also demonstrated the feasibility of designing drugs that exploit disease-specific protein complexes.
Advances in cellular target engagement technologies, such as the NanoBRET® TE platform, now allow researchers to directly assess compound engagement to protein complexes in live cells, bridging the gap between biochemical screening and cellular functional outcomes. Integrating assays that reflect physiological protein complex formation into drug discovery workflows can enhance the relevance of the assay results, optimize lead compounds more precisely, and ultimately accelerate the development of more targeted and safer therapies.
References
- Malumbres, M. (2014) Cyclin-dependent kinases. Genome Biol. 15, 122.
- Wells, C.I. et al. (2020) Quantifying CDK inhibitor selectivity in live cells. Nat. Commun. 11, 2743.
- Khan, Z.M. et al. (2020) Structural basis for the action of the drug trametinib at KSR-bound MEK. Nature 588, 509-14.
- Hu, M. and Chen, X. (2024) A review of the known MTA-cooperative PRMT5 inhibitors. RSC Adv. 14(53), 39653–91.
- Marjon, K. et al. (2016) MTAP Deletions in Cancer Create Vulnerability to Targeting of the MAT2A/PRMT5/RIOK1 Axis. Cell Rep. 15, 574-87.
- Cottrell, K.M. et al. (2024) Discovery of TNG908: A Selective, Brain Penetrant, MTA-Cooperative PRMT5 Inhibitor That Is Synthetically Lethal with MTAP-Deleted Cancers. J. Med. Chem. 67(8), 6064-80.
- Sarvary, I. et al. (2025) From DNA-Encoded Library Screening to AM-9747: An MTA-Cooperative PRMT5 Inhibitor with Potent Oral In Vivo Efficacy. J. Med. Chem. 68(6), 6534-57.
- Engstrom, L.D. et al. (2023) MRTX1719 Is an MTA-Cooperative PRMT5 Inhibitor That Exhibits Synthetic Lethality in Preclinical Models and Patients with MTAP-Deleted Cancer. Cancer Discov. 13(11), 2412-31.
Latest posts by Promega (see all)
- When Cancer Research Depends on Quality RNA: Maxwell® RSC in the Lab - March 30, 2026
- Accelerating Drug Discovery at Grove Biopharma with MyGlo® and ProNect® - December 9, 2025
- From Mt. Fuji to the Lab Bench: A UW-Madison Student’s Summer in Japan - December 3, 2025