Data generated by scientific instruments and decisions based on that data depend on optimal instrument performance. Clinical assays rely on mass spectrometric (MS) data for accurate results so that correct health related results are gained and appropriate results-based decisions are made. However, there are no generally agreed upon tools nor performance standards for mass spectrometry. Furthermore, while several software tools exist that serve to assist with the analysis of instrument performance, a dedicated reagent software package has yet to be created. For optimal liquid chromatography (LC) performance, parameters like retention time, peak width and peak heightare typically reported. Commonly monitored MS parameters include mass accuracy, mass resolution, signal-to noise, sensitivity, limit of detection (LOD), limit of quantitation (LOQ) and dynamic range.
The 6 × 5 LC-MS/MS Peptide Reference Mix is designed for use in method development and optimization,and for routine liquid chromatography (LC) and mass spectrometry (MS) instrument performance monitoring. The product is a mixture of 30 peptides: 6 sets of 5 isotopologues of the same peptide sequence. The isotopologues (Figure 1) differ only by the number of stable, heavy-labeled amino acids incorporated into the sequence. The labeled amino acids consist of uniform 13C and 15N atoms. Each of the isotopologues is indistinguishable chemically and chromatographically. However, since they differ in mass, they are clearly resolved by mass spectrometry.
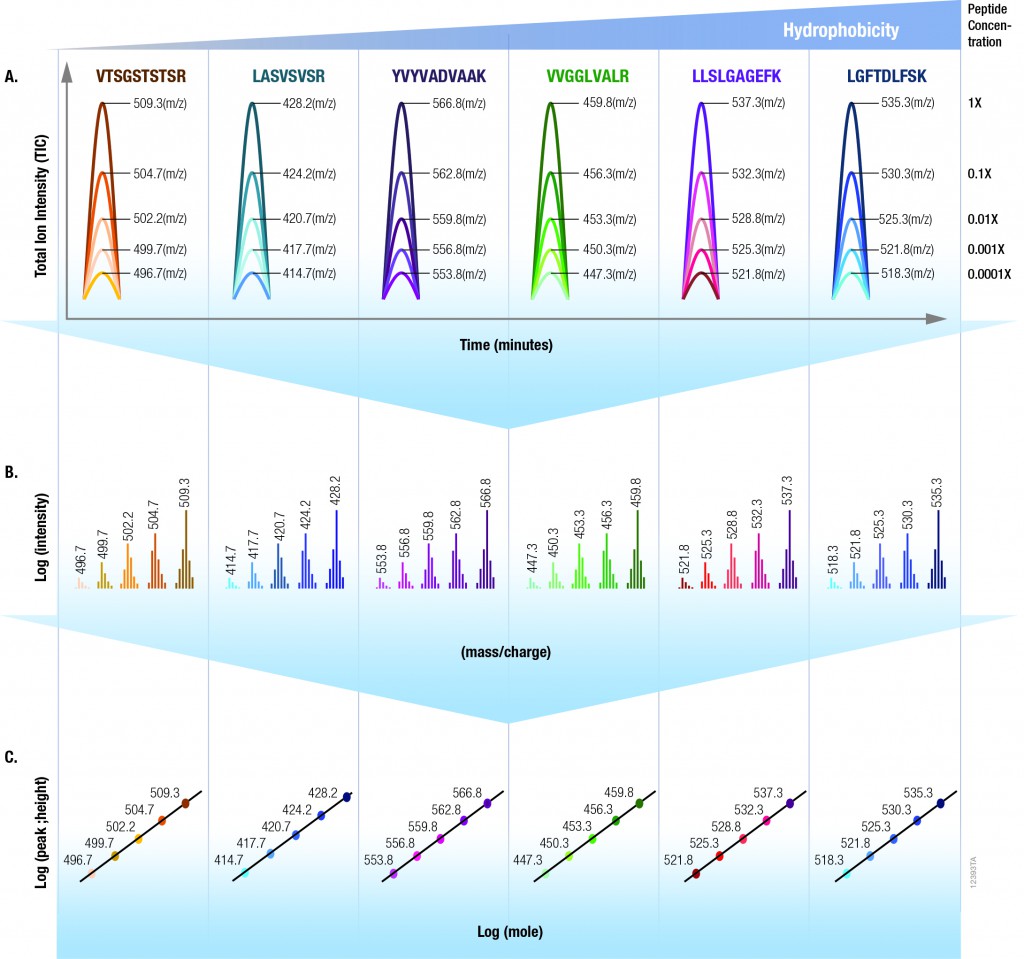
The isotopologues of each peptide are present in a series of tenfold differences in concentration or molar abundance. If 1pmol of the mixture is loaded onto an LC column, the next lighter isotopologue would be 100fmol, the next 10fmol, the second lightest 1fmol, and the lightest 100amol. This range allows assessment of the instrument’s dynamic range and sensitivity from a single run.
Peptides with a wide range of hydrophobicities were chosen to enable reporting of LC column performance. The most hydrophilic peptide gives users a tool to optimize the capture of hydrophilic peptides that might be difficult to capture otherwise, but that are too precious to use for method development.
To assist in data processing, a complementary software tool, is provided, the 6 × 5 LC-MS/MS Peptide Reference Mix Analysis Software (The PReMiS™ Software). The PReMiS™ Software produces a tabular report of calculated instrument parameters, graphical analysis of linearity curves as well as reporting the history of user-selected parameters such as LC retention time, peak height and mass accuracy. If the laboratory has a collection of instruments, there is also an option to compare parameters across instruments.