Transcriptional activation of genes within the nucleus of eukaryotic cells occurs by a variety of mechanisms. Typically, these mechanisms rely on the interaction of regulatory proteins (transcriptional activators or repressors) with specific DNA sequences that control gene expression. Upon DNA binding, regulatory proteins also interact with other proteins that are part of the RNA polymerase II transcriptional complex.
One type of transcriptional activation relies on inducing a conformational change in chromatin, the DNA-protein complex that makes up each chromosome within a cell. In a broad sense, “extended” or loosely wound chromatin is more accessible to transcription factors and can signify an actively transcribed gene. In contrast, “condensed” chromatin hinders access to transcription factors and is characteristic of a transcriptionally inactive state. Acetylation of lysine residues in histones—the primary constituents of the chromatin backbone—results in opening up the chromatin and consequent gene activation. Disruption of histone acetylation pathways is implicated in many types of cancer (1).
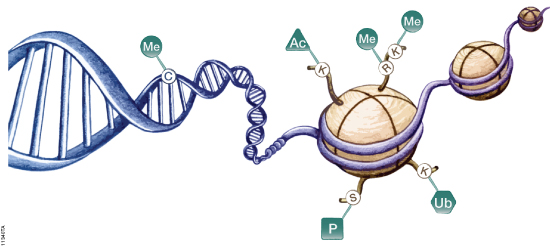
DNA is organized by protein:DNA complexes called nucleosomes in eukaryotes. Nucleosomes are composed of 147 base pairs of DNA wrapped around a histone octamer containing two copies of each core histone protein. Histone proteins play significant roles in many nuclear processes including transcription, DNA damage repair and heterochromatin formation. Histone proteins are extensively and dynamically post-translationally modified, and these post-translational modifications (PTMs) are thought to comprise a specific combinatorial PTM profile of a histone that dictates its specific function. Abnormal regulations of PTM may lead to developmental disorders and disease development such as cancer.
The ability to isolate and assay circulating cell-free DNA from plasma holds promise for improved diagnostics and treatment in the clinic. The use of blood-based non-invasive prenatal testing (NIPT) has been well described. Such testing is based on circulating cell-free fetal DNA in blood of a pregnant woman for diagnosis and screening of chromosomal anueploidy (e.g. Trisomy 21, Down Syndrome), sex-linked diseases, and genetic diseases that are known to result from a specific mutation in a single gene (1). Additionally, most cancers carry somatic mutations that are unique to the tumors, and dying tumor cells release small pieces of their DNA into the blood stream (2). This circulating cell-free tumor DNA can be used as a biomarker to “follow” cancer progression or regression during treatment, and diagnostic methods also are being developed to detect even early stage cancers from circulating tumor DNA (3). Further, increases in circulating cell-free DNA have been well documented after intense exercise, trauma, sepsis and even associated with autoimmune diseases such as system lupus erythematosus (SLE; 1,4). In these latter examples increases in extracellular DNA are associated with evolutionarily conserved innate immune responses involving the production of neutrophil extracellular traps (NETs). Monitoring the circulating cell-free DNA of NETs has implications for treatment and diagnosis of autoimmune diseases, cardiovascular events and traumatic injuries (4–7).
How Neutrophils Weave a Defensive Web
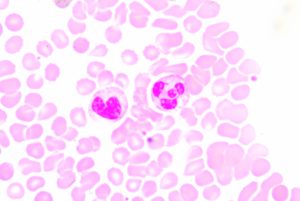
Blood smear showing two prominent neutrophils in the field of view
Neutrophils are the most abundant type of white blood cell and are part of the innate immune response, participating in non-specific immune responses to injury or pathogens. They are one of three types of granuolcytes, and can be recognized by their multi-lobed nucleus and the prominent granules that fill their cytoplasm. Generally they are first to the scene of injury or infection. Early in my scientific career, I was taught that neutrophils fought disease via phagocytosis and occasionally by firing a barrage of toxic enzymes and molecules at invaders. Mostly though they released cytokines that recruited the “important” cells of the specific immune system to the area.
For these reasons, I never really thought much about neutrophils. That is until recently, when I learned about Neutrophil Extracellular Traps (NETs). It turns out that neutrophils are pretty awesome, sacrificing themselves in a cloud-like explosion of DNA, chromatin, and granule proteins
XWe use cookies and similar technologies to make our website work, run analytics, improve our website, and show you personalized content and advertising. Some of these cookies are essential for our website to work. For others, we won’t set them unless you accept them. To learn more about our approach to Privacy we invite you to Read More
By clicking “Accept All”, you consent to the use of ALL the cookies. However you may visit Cookie Settings to provide a controlled consent.
We use cookies and similar technologies to make our website work, run analytics, improve our website, and show you personalized content and advertising. Some of these cookies are essential for our website to work. For others, we won’t set them unless you accept them. To find out more about cookies and how to manage cookies, read our Cookie Policy.
If you are located in the EEA, the United Kingdom, or Switzerland, you can change your settings at any time by clicking Manage Cookie Consent in the footer of our website.
Necessary cookies are absolutely essential for the website to function properly. These cookies ensure basic functionalities and security features of the website, anonymously.
Cookie
Duration
Description
cookielawinfo-checbox-analytics
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Analytics".
cookielawinfo-checbox-functional
11 months
The cookie is set by GDPR cookie consent to record the user consent for the cookies in the category "Functional".
cookielawinfo-checbox-others
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Other.
cookielawinfo-checkbox-advertisement
1 year
The cookie is set by GDPR cookie consent to record the user consent for the cookies in the category "Advertisement".
cookielawinfo-checkbox-necessary
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookies is used to store the user consent for the cookies in the category "Necessary".
cookielawinfo-checkbox-performance
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Performance".
gdpr_status
6 months 2 days
This cookie is set by the provider Media.net. This cookie is used to check the status whether the user has accepted the cookie consent box. It also helps in not showing the cookie consent box upon re-entry to the website.
lang
This cookie is used to store the language preferences of a user to serve up content in that stored language the next time user visit the website.
viewed_cookie_policy
11 months
The cookie is set by the GDPR Cookie Consent plugin and is used to store whether or not user has consented to the use of cookies. It does not store any personal data.
Analytical cookies are used to understand how visitors interact with the website. These cookies help provide information on metrics the number of visitors, bounce rate, traffic source, etc.
Cookie
Duration
Description
SC_ANALYTICS_GLOBAL_COOKIE
10 years
This cookie is associated with Sitecore content and personalization. This cookie is used to identify the repeat visit from a single user. Sitecore will send a persistent session cookie to the web client.
vuid
2 years
This domain of this cookie is owned by Vimeo. This cookie is used by vimeo to collect tracking information. It sets a unique ID to embed videos to the website.
WMF-Last-Access
1 month 18 hours 24 minutes
This cookie is used to calculate unique devices accessing the website.
_ga
2 years
This cookie is installed by Google Analytics. The cookie is used to calculate visitor, session, campaign data and keep track of site usage for the site's analytics report. The cookies store information anonymously and assign a randomly generated number to identify unique visitors.
_gid
1 day
This cookie is installed by Google Analytics. The cookie is used to store information of how visitors use a website and helps in creating an analytics report of how the website is doing. The data collected including the number visitors, the source where they have come from, and the pages visted in an anonymous form.
Advertisement cookies are used to provide visitors with relevant ads and marketing campaigns. These cookies track visitors across websites and collect information to provide customized ads.
Cookie
Duration
Description
IDE
1 year 24 days
Used by Google DoubleClick and stores information about how the user uses the website and any other advertisement before visiting the website. This is used to present users with ads that are relevant to them according to the user profile.
test_cookie
15 minutes
This cookie is set by doubleclick.net. The purpose of the cookie is to determine if the user's browser supports cookies.
VISITOR_INFO1_LIVE
5 months 27 days
This cookie is set by Youtube. Used to track the information of the embedded YouTube videos on a website.
Performance cookies are used to understand and analyze the key performance indexes of the website which helps in delivering a better user experience for the visitors.
Cookie
Duration
Description
YSC
session
This cookies is set by Youtube and is used to track the views of embedded videos.
_gat_UA-62336821-1
1 minute
This is a pattern type cookie set by Google Analytics, where the pattern element on the name contains the unique identity number of the account or website it relates to. It appears to be a variation of the _gat cookie which is used to limit the amount of data recorded by Google on high traffic volume websites.