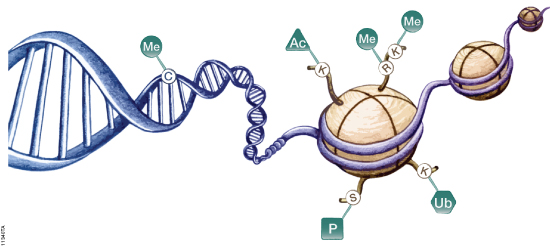
DNA is organized by protein:DNA complexes called nucleosomes in eukaryotes. Nucleosomes are composed of 147 base pairs of DNA wrapped around a histone octamer containing two copies of each core histone protein. Histone proteins play significant roles in many nuclear processes including transcription, DNA damage repair and heterochromatin formation. Histone proteins are extensively and dynamically post-translationally modified, and these post-translational modifications (PTMs) are thought to comprise a specific combinatorial PTM profile of a histone that dictates its specific function. Abnormal regulations of PTM may lead to developmental disorders and disease development such as cancer.
Continue reading “Mass Spec Analysis of PTMs Using Minimal Sample Material”