The use of mass spectrometry for the characterization of individual or complex protein samples continues to be one of the fastest growing fields in the life science market.
Bottom-up proteomics is the traditional approach to address these questions. Optimization of each the individual steps (e.g. sample prep, digestion and instrument performance) is critical to the overall success of the entire experiment.
To address issues that may arise in your experimental design, Promega has developed unique tools and complementary webinars to help you along the way.
Here you can find a summary of individual webinars for the following topics:
Data generated by scientific instruments and decisions based on that data depend on optimal instrument performance. Clinical assays rely on mass spectrometric (MS) data for accurate results so that correct health related results are gained and appropriate results-based decisions are made. However, there are no generally agreed upon tools nor performance standards for mass spectrometry. Furthermore, while several software tools exist that serve to assist with the analysis of instrument performance, a dedicated reagent software package has yet to be created. For optimal liquid chromatography (LC) performance, parameters like retention time, peak width and peak heightare typically reported. Commonly monitored MS parameters include mass accuracy, mass resolution, signal-to noise, sensitivity, limit of detection (LOD), limit of quantitation (LOQ) and dynamic range.
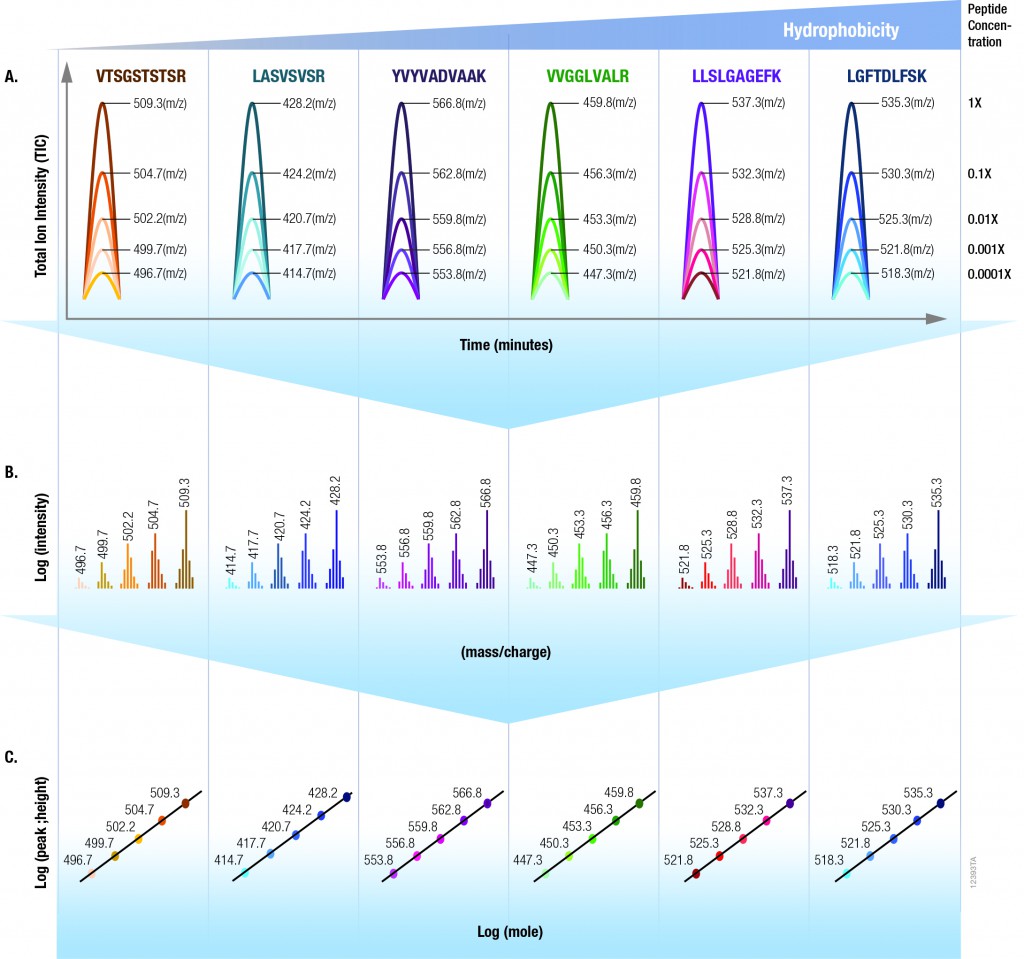
The 6 × 5 LC-MS/MS Peptide Reference Mix is designed for use in method development and optimization,and for routine liquid chromatography (LC) and mass spectrometry (MS) instrument performance monitoring. The product is a mixture of 30 peptides: 6 sets of 5 isotopologues of the same peptide sequence. The isotopologues (Figure 1) differ only by the number of stable, heavy-labeled amino acids incorporated into the sequence. The labeled amino acids consist of uniform 13C and 15N atoms. Each of the isotopologues is indistinguishable chemically and chromatographically. However, since they differ in mass, they are clearly resolved by mass spectrometry.
Figure 1.
The isotopologues of each peptide are present in a series of tenfold differences in concentration or molar abundance. If 1pmol of the mixture is loaded onto an LC column, the next lighter isotopologue would be 100fmol, the next 10fmol, the second lightest 1fmol, and the lightest 100amol. This range allows assessment of the instrument’s dynamic range and sensitivity from a single run.
Peptides with a wide range of hydrophobicities were chosen to enable reporting of LC column performance. The most hydrophilic peptide gives users a tool to optimize the capture of hydrophilic peptides that might be difficult to capture otherwise, but that are too precious to use for method development.
To assist in data processing, a complementary software tool, is provided, the 6 × 5 LC-MS/MS Peptide Reference Mix Analysis Software (The PReMiS™ Software). The PReMiS™ Software produces a tabular report of calculated instrument parameters, graphical analysis of linearity curves as well as reporting the history of user-selected parameters such as LC retention time, peak height and mass accuracy. If the laboratory has a collection of instruments, there is also an option to compare parameters across instruments.
XWe use cookies and similar technologies to make our website work, run analytics, improve our website, and show you personalized content and advertising. Some of these cookies are essential for our website to work. For others, we won’t set them unless you accept them. To learn more about our approach to Privacy we invite you to Read More
By clicking “Accept All”, you consent to the use of ALL the cookies. However you may visit Cookie Settings to provide a controlled consent.
We use cookies and similar technologies to make our website work, run analytics, improve our website, and show you personalized content and advertising. Some of these cookies are essential for our website to work. For others, we won’t set them unless you accept them. To find out more about cookies and how to manage cookies, read our Cookie Policy.
If you are located in the EEA, the United Kingdom, or Switzerland, you can change your settings at any time by clicking Manage Cookie Consent in the footer of our website.
Necessary cookies are absolutely essential for the website to function properly. These cookies ensure basic functionalities and security features of the website, anonymously.
Cookie
Duration
Description
cookielawinfo-checbox-analytics
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Analytics".
cookielawinfo-checbox-functional
11 months
The cookie is set by GDPR cookie consent to record the user consent for the cookies in the category "Functional".
cookielawinfo-checbox-others
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Other.
cookielawinfo-checkbox-advertisement
1 year
The cookie is set by GDPR cookie consent to record the user consent for the cookies in the category "Advertisement".
cookielawinfo-checkbox-necessary
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookies is used to store the user consent for the cookies in the category "Necessary".
cookielawinfo-checkbox-performance
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Performance".
gdpr_status
6 months 2 days
This cookie is set by the provider Media.net. This cookie is used to check the status whether the user has accepted the cookie consent box. It also helps in not showing the cookie consent box upon re-entry to the website.
lang
This cookie is used to store the language preferences of a user to serve up content in that stored language the next time user visit the website.
viewed_cookie_policy
11 months
The cookie is set by the GDPR Cookie Consent plugin and is used to store whether or not user has consented to the use of cookies. It does not store any personal data.
Analytical cookies are used to understand how visitors interact with the website. These cookies help provide information on metrics the number of visitors, bounce rate, traffic source, etc.
Cookie
Duration
Description
SC_ANALYTICS_GLOBAL_COOKIE
10 years
This cookie is associated with Sitecore content and personalization. This cookie is used to identify the repeat visit from a single user. Sitecore will send a persistent session cookie to the web client.
vuid
2 years
This domain of this cookie is owned by Vimeo. This cookie is used by vimeo to collect tracking information. It sets a unique ID to embed videos to the website.
WMF-Last-Access
1 month 18 hours 24 minutes
This cookie is used to calculate unique devices accessing the website.
_ga
2 years
This cookie is installed by Google Analytics. The cookie is used to calculate visitor, session, campaign data and keep track of site usage for the site's analytics report. The cookies store information anonymously and assign a randomly generated number to identify unique visitors.
_gid
1 day
This cookie is installed by Google Analytics. The cookie is used to store information of how visitors use a website and helps in creating an analytics report of how the website is doing. The data collected including the number visitors, the source where they have come from, and the pages visted in an anonymous form.
Advertisement cookies are used to provide visitors with relevant ads and marketing campaigns. These cookies track visitors across websites and collect information to provide customized ads.
Cookie
Duration
Description
IDE
1 year 24 days
Used by Google DoubleClick and stores information about how the user uses the website and any other advertisement before visiting the website. This is used to present users with ads that are relevant to them according to the user profile.
test_cookie
15 minutes
This cookie is set by doubleclick.net. The purpose of the cookie is to determine if the user's browser supports cookies.
VISITOR_INFO1_LIVE
5 months 27 days
This cookie is set by Youtube. Used to track the information of the embedded YouTube videos on a website.
Performance cookies are used to understand and analyze the key performance indexes of the website which helps in delivering a better user experience for the visitors.
Cookie
Duration
Description
YSC
session
This cookies is set by Youtube and is used to track the views of embedded videos.
_gat_UA-62336821-1
1 minute
This is a pattern type cookie set by Google Analytics, where the pattern element on the name contains the unique identity number of the account or website it relates to. It appears to be a variation of the _gat cookie which is used to limit the amount of data recorded by Google on high traffic volume websites.