Studying protein function in live cells is limited by the tools available to analyze the expression and interactions of those proteins. Although mass spectrometry and antibody-based protein detection are valuable technologies for protein analysis, both methods have drawbacks that limit the range of targets and contexts in which proteins can be investigated.
Mass spectrometry is often poor at detecting low-abundance proteins. Antibody-based techniques require high quality, specific antibodies, which can be difficult to impossible to acquire. Both methods require cell lysis, preventing real-time analysis and limiting the physiological relevance, and both methods can be limiting for higher-throughput analysis. While plasmid-based overexpression of tagged target proteins simplifies detection and can allow for real time analysis, protein levels don’t typically resemble endogenous levels. Overexpression also has the potential to create experimental artifacts or limit the dynamic range of an observed response.
In 2018, Promega R&D scientists published a paper in ACS Chemical Biology demonstrating the use of CRISPR/Cas9 to integrate the 11 amino acid, bioluminescent HiBiT tag directly into the genome to serve as an easily measured reporter for endogenous proteins. This provides a highly quantitative method for investigating cellular protein dynamics that sidesteps the need for cloning and other drawbacks to conventional methods, including the ability to measure changing protein dynamics in real-time. (For more details about CRISPR/Cas9 knock-in tagging and other applications, read this blog.)
While their findings showed that this method provides efficient and specific tagging of endogenous proteins, the research was limited to just five different proteins within a single signaling pathway in two cell lines. This left unanswered questions about whether this approach was scalable, had broader applications and how accurately the natural biology of the cells was represented.
Continue reading “CRISPR/Cas9 HiBiT Knock-In: A Scalable Approach for Studying Endogenous Protein Dynamics”

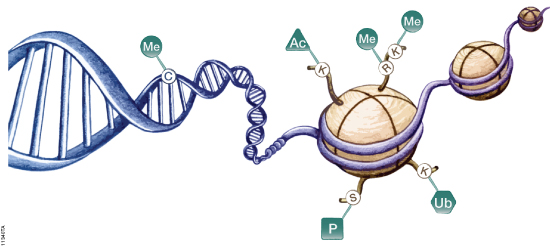

 Protein phosphorylation is a very important protein post-translational modification that controls many cellular processes including metabolism, transcriptional and translation regulation, degradation of proteins, cellular signaling and communication, proliferation, differentiation, and cell survival (1). Approximately 35% of human proteins are phosphorylated. Phosphoproteins are low in abundance, and, therefore, are challenging to detect and characterize by mass spectrometry. Different enrichment systems have been developed to isolate phosphopeptides. Among these techniques, immobilized metal affinity chromatography (IMAC) using Fe3+ and Ga3+ has been widely used for the enrichment of phosphopeptides.
Protein phosphorylation is a very important protein post-translational modification that controls many cellular processes including metabolism, transcriptional and translation regulation, degradation of proteins, cellular signaling and communication, proliferation, differentiation, and cell survival (1). Approximately 35% of human proteins are phosphorylated. Phosphoproteins are low in abundance, and, therefore, are challenging to detect and characterize by mass spectrometry. Different enrichment systems have been developed to isolate phosphopeptides. Among these techniques, immobilized metal affinity chromatography (IMAC) using Fe3+ and Ga3+ has been widely used for the enrichment of phosphopeptides.