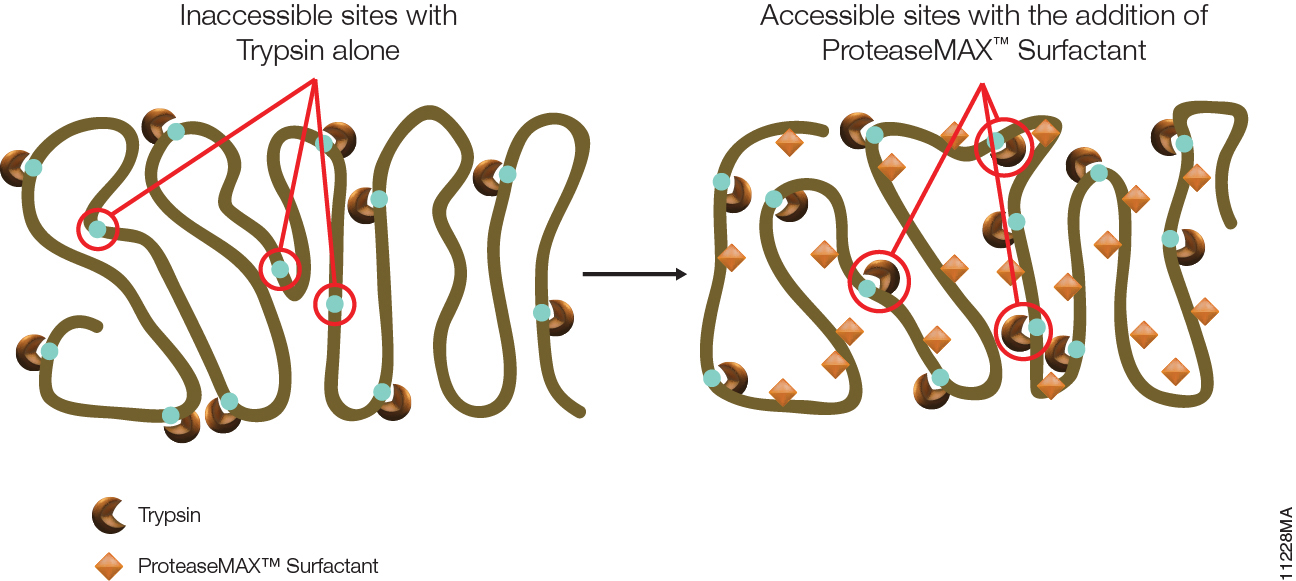
Proteomics, the analysis of the entire protein content of a living system, has become a vital part of life science research, and mass spectrometry (MS) is the method for analyzing proteins. MS analysis of protein content allows researchers to identify proteins, sequence them and determine the nature of post translational modifications.
Mass spectrometry allows characterization of molecules by converting them to ions so that they can be manipulated in electrical and magnetic fields. Basically a small sample (analyte) is ionized, usually to cations by loss of an electron. After ionization, the charged particles (ions) are separated by mass and charge; the separated particles are measured and data displayed as a mass spectrum. The mass spectrum is typically presented as a bar graph where each peak represents a single charged particle having a specific mass-to-charge (m/z) ratio. The height of the peak represents the relative abundance of the particle. The number and relative abundance of the ions reveal how different parts of the molecule relate to each other.
For the study of large, organic macromolecules, matrix associated laser desorption/ionization (MALDI) or tandem mass spec/collision induced dissociation (MS/MS) techniques are often used to generate the charged particles from the analyte. MS analysis brings sensitivity and specificity to proteome analysis. The technique has excellent resolution and is able to distinguish one ion from another, even when their m/z ratios are similar. Macromolecules are present in extremely different concentrations in the cells, and MS analysis can detect biomolecules across five logs of concentration.
Continue reading “Monitoring Mass Spec Instrument Performance and Sample Preparation”