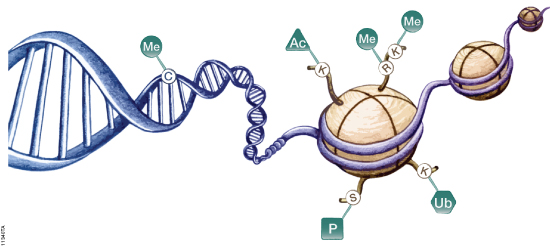
DNA is organized by protein:DNA complexes called nucleosomes in eukaryotes. Nucleosomes are composed of 147 base pairs of DNA wrapped around a histone octamer containing two copies of each core histone protein. Histone proteins play significant roles in many nuclear processes including transcription, DNA damage repair and heterochromatin formation. Histone proteins are extensively and dynamically post-translationally modified, and these post-translational modifications (PTMs) are thought to comprise a specific combinatorial PTM profile of a histone that dictates its specific function. Abnormal regulations of PTM may lead to developmental disorders and disease development such as cancer.
Antibodies have been widely used to characterize histones and histone PTMs. However, antibody-based techniques have several limitations. Mass spectrometry (MS) has therefore emerged as the most suitable analytical tool to quantify proteomes and protein PTMs. The most commonly used strategy is still bottom-up MS, and the most widely adopted protocol includes derivatization of lysine residues in histones to allow trypsin to generate Arg-C like peptides (4–20 aa). However, samples such as primary tissues, complex model systems, and biofluids are hard to retrieve in large quantities. Because of this, it is critical to know whether the amount of sample available would lead to an exhaustive analysis if subjected to MS.
In a recent publication, Guo, et al. examined (1) the reproducibility in quantification of histone PTMs using a wide range of starting material: from 50,000 to 5,000,000 cells. They used four different cell lines: HeLa, 293T, human embryonic stem cells (hESCs), and myoblasts. Their results demonstrated that accurate quantification of abundant histone PTMs can be efficiently obtained by using low-resolution MS and as low as 50,000 cells as starting material Low abundance histone marks showed more variability in quantification when comparing different amounts of starting material, so a larger amount of starting material (at least 500,000 cells) is recommended.
Reference
Guo, Q. et al. (2017) Assessment of Quantification Precision of Histone Post-Translational Modifications by Using an Ion Trap and down To 50,000 Cells as Starting Material. J. Proteome Res. 17, 234–42.
Try a sample of high-efficiency Trypsin Platinum today!
Visit our website for more on Trypsin Platinum, Mass Spectrometry Grade, with enhanced proteolytic efficiency and superior autoproteolytic resistance.